Model Optimization and In Silico Analysis of Potential Dipeptidyl Peptidase IV Antagonists from GC-MS Identified Compounds in Nauclea latifolia Leaf Extracts




Abstract
:1. Introduction
2. Results
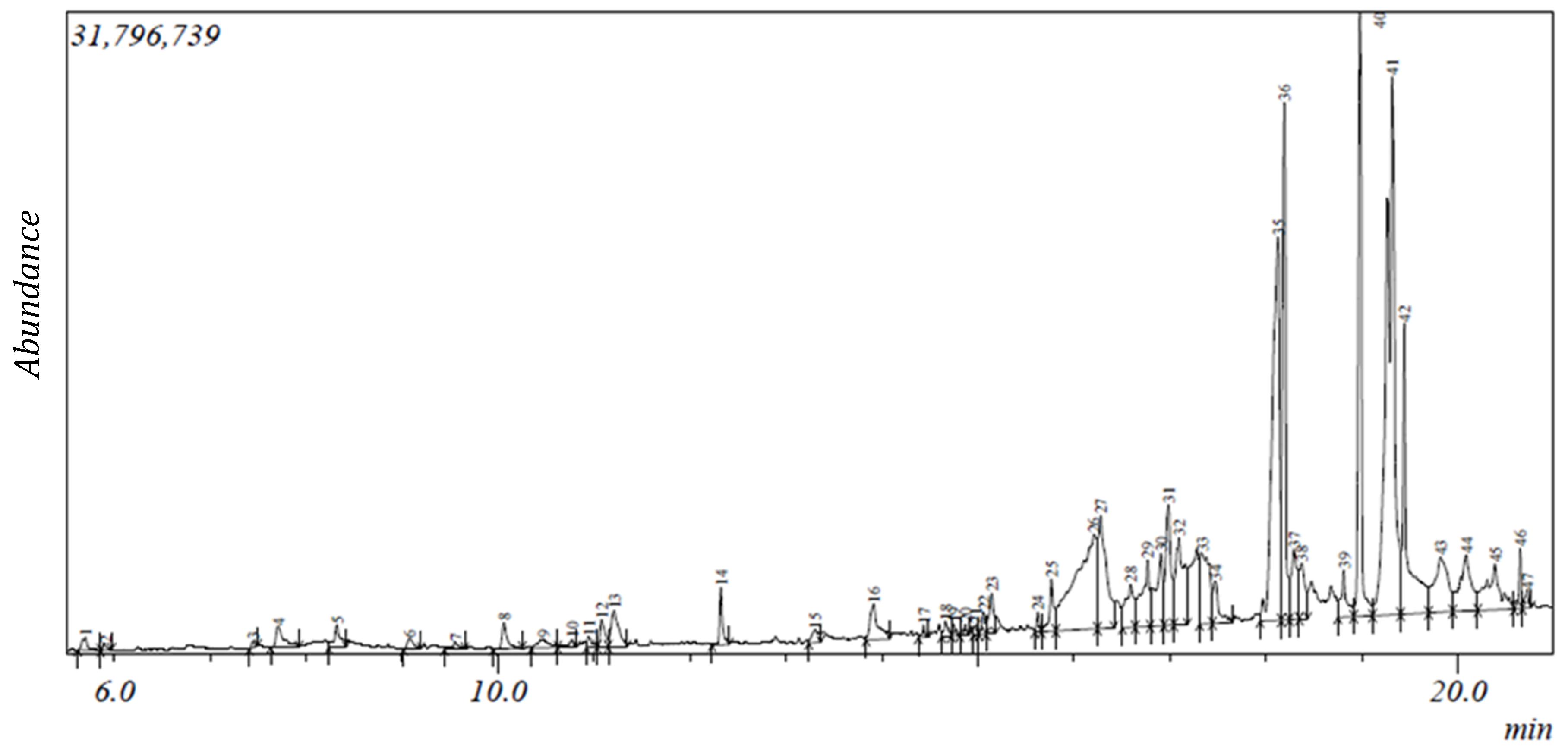
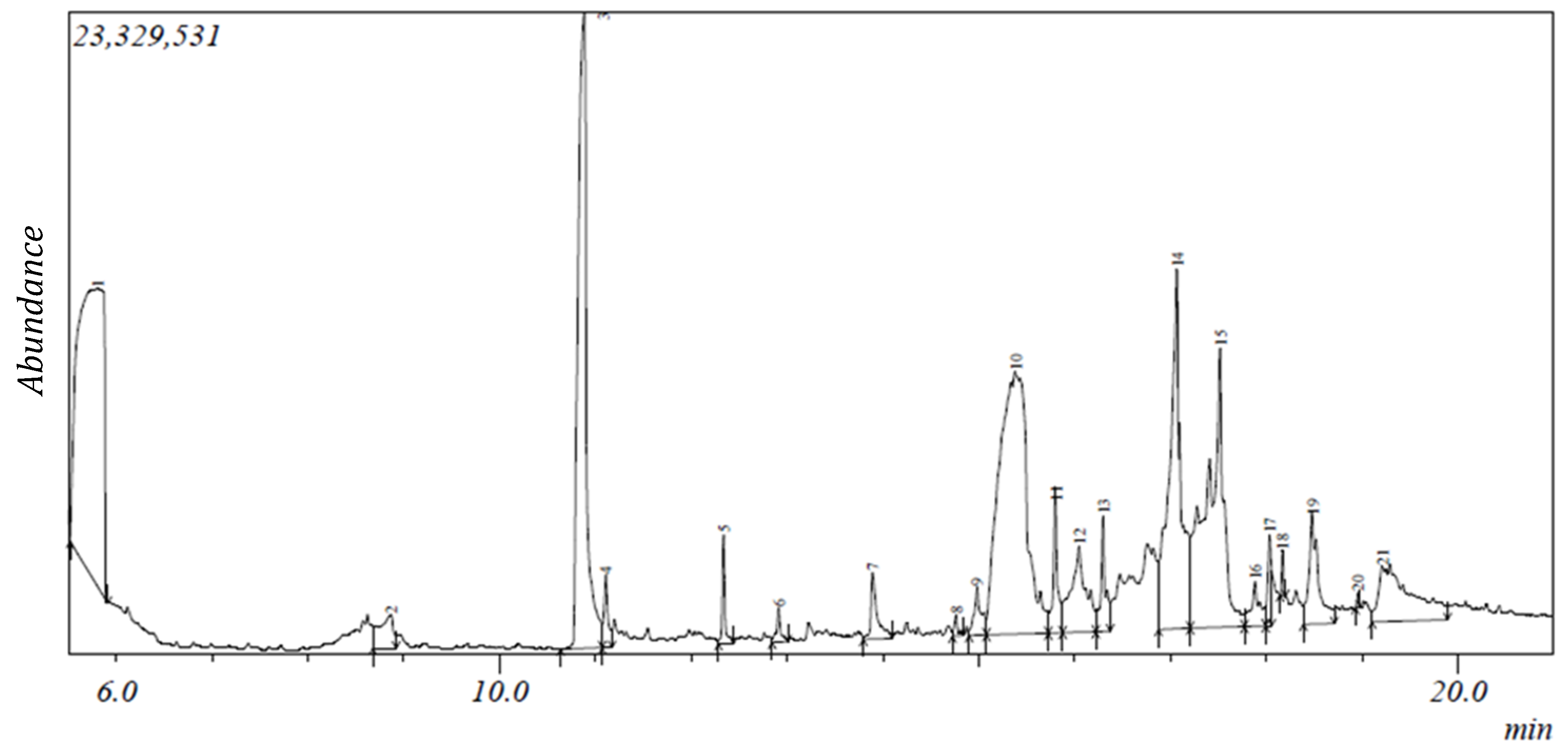
2.1. Gas Chromatography-Mass Spectroscopy (GC-MS) Results
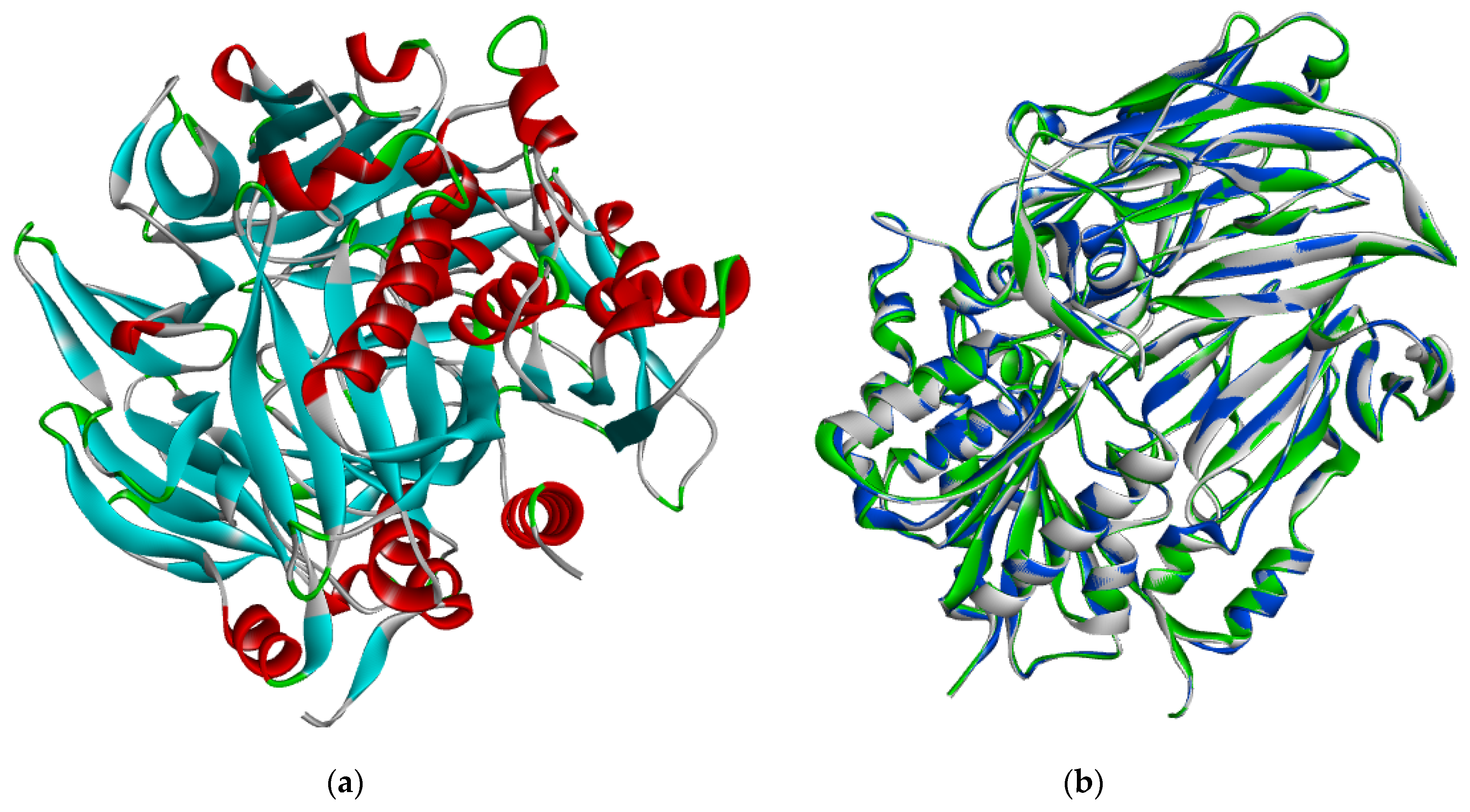
2.2. Protein Sequence Retrieval and Model Optimization Results
2.3. Energy Minimization, Physicochemical Analysis and Model Evaluation Results
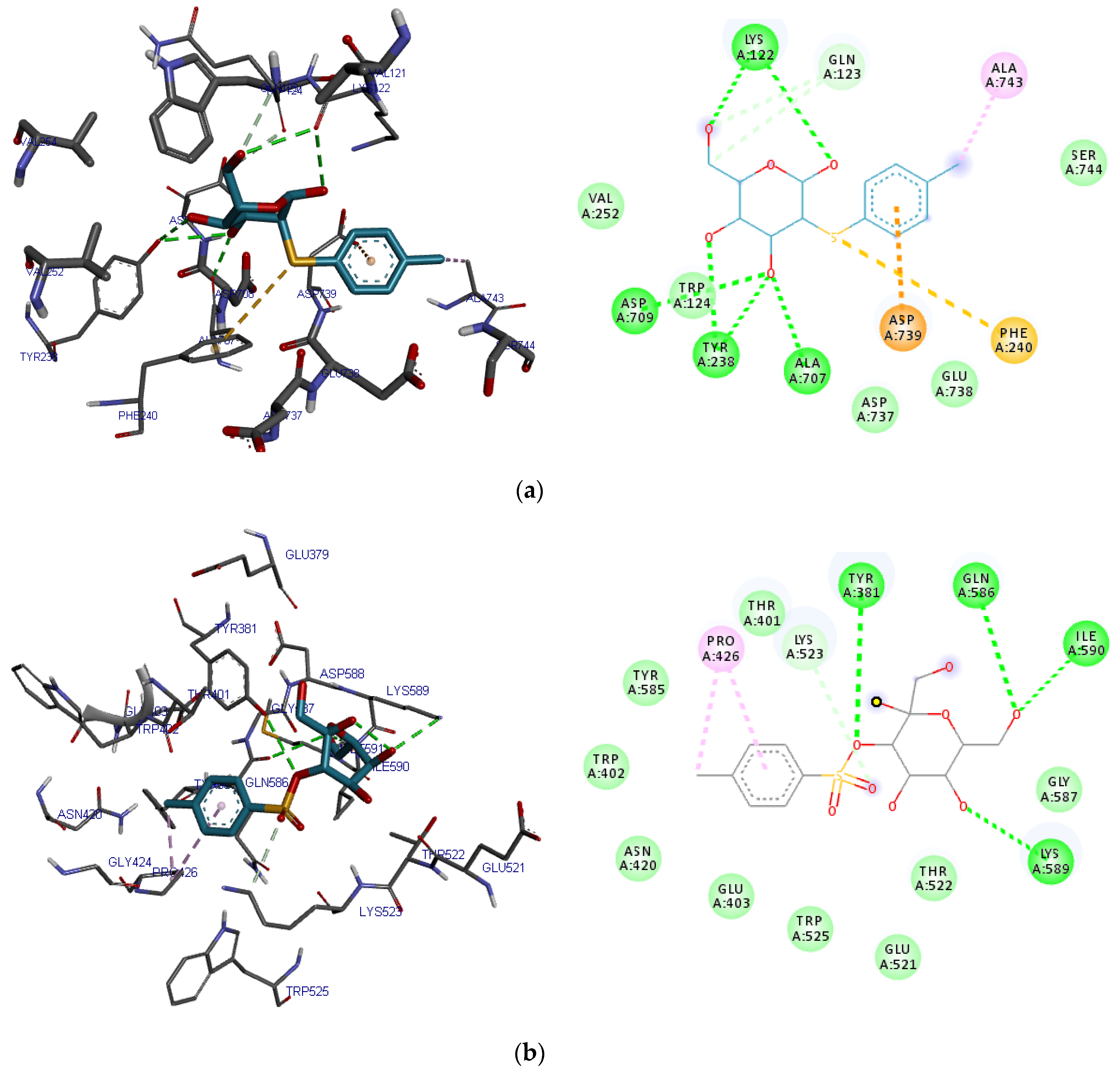
2.4. Pocket Identification and Molecular Docking Simulation Results
2.5. Molecular Docking Simulation Results
2.6. Druglikeness, Pharmacokinetic and Toxicity Prediction
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Plant Collection, Identification and Extract Preparation
4.3. GC-MS Analysis
4.4. Ligand Modeling
4.5. Protein Sequence Retrieval, Model Optimization and Energy Minimization
4.6. Model Evaluation, Physicochemical Analysis and Pocket Identification
4.7. Molecular Docking Simulation
4.8. Druglikeness, Pharmacokinetic and Toxicity Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kabel, A.M. Zinc/alogliptin combination attenuates testicular toxicity induced by doxorubicin in rats: Role of oxidative stress, apoptosis and TGF-β1/NF-κB signalling. Biomed. Pharmacother. 2018, 97, 439–449. [Google Scholar] [CrossRef]
- Knop, F.K.; Aaboe, K.; Vilsbøll, T.; Vølund, A.; Holst, J.J.; Krarup, T.; Madsbad, S. Impaired incretin effect and fasting hyperglucagonaemia characterizing type 2 diabetic subjects are early signs of dysmetabolism in obesity. Diabetes Obes. Metab. 2012, 14, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Kabel, A.M.; Omar, M.S.; Alhadhrami, A.; Alharthi, S.S.; Alrobaian, M.M. Linagliptin potentiates the effect of L-dopa on the behavioural, biochemical and immunohistochemical changes in experimentally-induced Parkinsonism: Role of toll-like receptor 4, TGF-β1, NF-κB and glucagon-like peptide 1. Physiol. Behav. 2018, 188, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F.; Holst, J.J. Dipeptidyl peptidase IV inhibitors: A promising new therapeutic approach for the management of type 2 diabetes. Int. J. Biochem. Cell Biol. 2006, 38, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagonlike peptide-1 receptor agonists and dipeptidyl peptidase-4. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Koska, J.; Sands, M.; Burciu, C.; Reaven, P. Cardiovascular effects of dipeptidyl peptidase-4 inhibitors in patients with type 2 diabetes. Diab. Vasc. Dis. Res. 2015, 12, 154–163. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Gupta, A.; Al-Aubaidy, H.A.; Mohammed, B.I. Glucose dependent insulinotropic polypeptide and dipeptidyl peptidase inhibitors: Their roles in management of type 2 diabetes mellitus. Diabetes Metab. Syndr. 2016, 10, S170–S175. [Google Scholar] [CrossRef]
- Hopsu-Havu, V.K.; Glenner, G.G. A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-β-naphthylamide. Histochemie 1966, 7, 197–201. [Google Scholar] [CrossRef]
- Lacroix, I.M.; Li-Chan, E.C. Food-derived dipeptidyl-peptidase IV inhibitors as a potential approach for glycemic regulation–current knowledge and future research considerations. Trends Food Sci. Technol. 2016, 54, 1–16. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.; Shi, X.; Gu, W. Dipeptidyl peptidase IV (DPP IV): A novel emerging target for the treatment of type 2 diabetes. J. Nanjing Med. Univ. 2009, 23, 228–235. [Google Scholar] [CrossRef]
- Mulvilhill, E.E. Dipeptidyl peptidase inhibitor therapy in type 2 diabetes: Control of the incretin axis and regulation of postprandial glucose and lipid metabolism. Peptides 2018, 100, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Abbas, G.; Hussain, H.; Hamaed, A.; Supuran, C.T. The management of diabetes mellitus-imperative role of natural products against dipeptidyl peptidase-4, α-glucosidase and sodium-dependent glucose co-transporter 2 (SGLT2). Bioorganic Chem. 2019, 86, 305–315. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, C.H.; Demuth, H.U.; Kim, S.J.; Pospisilik, J.A.; Pederson, R.A. Applications of dipeptidyl peptidase IV inhibitors in diabetes mellitus. Int. J. Biochem. Cell Biol. 2006, 38, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef]
- Leung, M.; Leung, D.Y.; Wong, V.W. Effects of dipeptidyl peptidase-4 inhibitors on cardiac and endothelial function in type 2 diabetes mellitus: A pilot study. Diab. Vasc. Dis. Res. 2016, 13, 236–243. [Google Scholar] [CrossRef]
- Ala’a, G.A.; Alkhaldi, E.H.; Alzahrani, A.S.; Alghamdi, A.K.; Alghamdi, W.Y.; Kabel, A.M. Dipeptidyl peptidase-4 inhibitors: Anti-diabetic drugs with potential effects on cancer. Diabetes Metab. Syndr. 2018, 13, 36–39. [Google Scholar]
- Demuth, H.U.; McIntosh, C.H.; Pederson, R.A. Type 2 diabetes-therapy with dipeptidyl peptidase IV inhibitors. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 33–44. [Google Scholar] [CrossRef]
- Nabeno, M.; Akahoshi, F.; Kishida, H.; Miyaguchi, I.; Tanaka, Y.; Ishii, S.; Kadowaki, T. A comparative study of the binding modes of recently launched dipeptidyl peptidase IV inhibitors in the active site. Biochem. Biophys. Res. Commun. 2013, 434, 191–196. [Google Scholar] [CrossRef]
- Karagiannis, T.; Boura, P.; Tsapas, A. Safety of dipeptidyl peptidase 4 inhibitors: A perspective review. Ther. Adv. Drug Saf. 2014, 5, 138–146. [Google Scholar] [CrossRef]
- Boucherle, B.; Haudecoeur, R.; Queiroz, E.F.; De Waard, M.; Wolfender, J.L.; Robins, R.J.; Boumendjel, A. Nauclea latifolia: Biological activity and alkaloid phytochemistry of a West African tree. Nat. Prod. Rep. 2016, 33, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Antia, B.S.; Okokon, J.E. Phytochemical composition and antidiabetic activity of ethanol root extract of Nauclea latifolia. J. Phytopharmacol. 2014, 3, 52–56. [Google Scholar]
- Gidado, A.; Ameh, D.A.; Atawodi, S.E.; Ibrahim, S. A preliminary study of the mechanism of hypoglycaemic activity of Nauclea latifolia leaf ethanolic extract. J. Complement. Integr. Med. 2012, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Effiong, G.S.; Mgbeje, B.I.; Igile, G.O.; Atangwho, J.I.; Eyong, E.U.; Ebong, P.E. Antioxidant enzymes activity and hormonal changes following administration of ethanolic leaves extracts of Nauclea latifolia and Gongronema latifolium in streptozotocin induced-diabetic rats. Eur. J. Med. Plants 2013, 3, 297–309. [Google Scholar] [CrossRef]
- Onyesom, I.; Osioma, E.; Okereke, P.C. Nauclea latifolia aqueous leaf extract eliminates hepatic and cerebral Plasmodium berghei parasite in experimental mice. Asian Pac. J. Trop. Biomed. 2015, 5, 546–551. [Google Scholar] [CrossRef]
- Seukep, J.A.; Sandjo, L.P.; Ngadjui, B.T.; Kuete, V. Antibacterial and antibiotic-resistance modifying activity of the extracts and compounds from Nauclea pobeguinii against Gram-negative multi-drug resistant phenotypes. BMC Complement. Altern. Med. 2016, 16, 193. [Google Scholar] [CrossRef]
- Donalisio, M.; Nana, H.M.; Ngane, R.A.; Gatsing, D.; Tchinda, A.T.; Rovito, R.; Cagno, V.; Cagliero, C.; Boyom, F.F.; Rubiolo, P.; et al. In vitro anti-Herpes simplex virus activity of crude extract of the roots of Nauclea latifolia Smith (Rubiaceae). BMC Complement. Altern. Med. 2013, 13, 266–274. [Google Scholar] [CrossRef]
- Balogun, M.E.; Jeje, S.O.; Salami, S.A.; Onwe, P.E.; Folawiyo, M.A. Anti-ulcerogenic and gastric anti-secretory effects of Nauclea latifolia extract in male albino rats. Eur. J. Exp. Biol. 2015, 5, 74–80. [Google Scholar]
- Taïwe, G.S.; Ngo Bum, E.; Talla, E.; Dimo, T.; Dawe, A.; Sinniger, V.; Bonaz, B.; Boumendjel, A.; De Waard, M. Nauclea latifolia Smith (Rubiaceae) exerts antinociceptive effects in neuropathic pain induced by chronic constriction injury of the sciatic nerve. J. Ethnopharmacol. 2014, 151, 445–451. [Google Scholar] [CrossRef]
- Nwaehujor, C.O.; Onyenweaku, E.O.; Ezeigbo, I.I.; Asuzu, O.V. Evaluation of the hepato-protective potential of Sarcocephalus latifolius leaf methanol extracts in paracetamol-induced liver damage of mice. Comp. Clin. Path. 2016, 25, 1053–1059. [Google Scholar] [CrossRef]
- Haudecoeur, R.; Peuchmaur, M.; Peres, B.; Rome, M.; Taiwe, G.S.; Boumendjel, A.; Boucherle, B. Traditional uses, phytochemistry and pharmacological properties of African Nauclea species: A review. J. Ethnopharmacol. 2018, 212, 106–136. [Google Scholar] [CrossRef] [PubMed]
- Kalhotra, P.; Chittepu, V.; Osorio-Revilla, G.; Gallardo-Velázquez, T. Structure–activity relationship and molecular docking of natural product library reveal chrysin as a novel dipeptidyl peptidase-4 (dpp-4) inhibitor: An integrated in silico and in vitro study. Molecules 2018, 23, 1368. [Google Scholar] [CrossRef] [PubMed]
- Sabiu, S.; Ashafa, A.O. Membrane stabilization and kinetics of carbohydrate metabolizing enzymes (α-amylase and α-glucosidase) inhibitory potentials of Eucalyptus obliqua L. Her. (Myrtaceae) Blakely ethanolic leaf extract: An in vitro assessment. S. Afr. J. Bot. 2016, 105, 264–269. [Google Scholar] [CrossRef]
- Ameachi, N.C.; Chijioke, C.L. Evaluation of bioactive compounds in Pseudarenthemum tunicatum leaves using gas chromatography-mass spectrometry. Sci. Paper Ser. 2018, 18, 53–59. [Google Scholar]
- Rajkumar, P.; Selvaraj, S.; Suganya, R.; Velmurugan, D.; Kumaresan, S. GC-MS characterization of the anti-diabetic compounds from the flowers of cassia auriculata (AVARAM): A structure based molecular docking studies. Int. J. Innov. Res. Sci. Eng. Technol. 2016, 1, 85–93. [Google Scholar]
- Adeoye-Isijola, M.O.; Olajuyigbe, O.O.; Jonathan, S.G.; Coopoosamy, R.M. Bioactive compounds in ethanol extract of Lentinus squarrosulus Mont-a Nigerian medicinal macrofungus. Afr. J. Tradit. Complement. Altern. Med. 2016, 15, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Adebayo, A.H.; Alade, A.B.; Yakubu, O.F. Gas chromatography-mass spectrometry analysis of Viburnum Opulus (L) extract and its toxicity studies in rats. Asian J. Pharm. Clin. Res. 2017, 10, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Ajiboye, B.O.; Ojo, O.A.; Adeyonu, O.; Imiere, O.; Olayide, I.; Fadaka, A.; Oyinloye, B.E. Inhibitory effect on key enzymes relevant to acute type-2 diabetes and antioxidative activity of ethanolic extract of Artocarpus heterophyllus stem bark. J. Acute Dis. 2016, 5, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Vedamurthy, G.V.; Ahmad, H.; Onteru, S.K.; Saxena, V.K. In silico homology modelling and prediction of novel epitopic peptides from P24 protein of Haemonchus contortus. Gene 2019, 703, 102–111. [Google Scholar] [CrossRef]
- Isa, M.A. Homology modeling and molecular dynamic simulation of UDP-N-acetylmuramoyl-l-alanine-d-glutamate ligase (MurD) from Mycobacterium tuberculosis H37Rv using in silico approach. Comput. Biol. Chem. 2019, 78, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Abi Hussein, H.; Geneix, C.; Petitjean, M.; Borrel, A.; Flatters, D.; Camproux, A.C. Global vision of druggability issues: Applications and perspectives. Drug Discov. Today 2017, 22, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.C. PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef] [Green Version]
- Iheagwam, F.N.; Ogunlana, O.O.; Ogunlana, O.E.; Isewon, I.; Oyelade, J. Potential anti-cancer flavonoids isolated from Caesalpinia bonduc young twigs and leaves: Molecular docking and in silico studies. Bioinform. Biol. Insights 2019, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.M.; Chen, C.C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins: Struct. Funct. Bioinf. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.; Denise, M.T.; O’connor, S.; Gorrell, M.D. Inhibitor selectivity in the clinical application of dipeptidyl peptidase-4 inhibition. Clin. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Metzler, W.J.; Yanchunas, J.; Weigelt, C.; Kish, K.; Klei, H.E.; Xie, D.; Zhang, Y.; Corbett, M.; Tamura, J.K.; He, B.; et al. Involvement of DPP-IV catalytic residues in enzyme–saxagliptin complex formation. Protein Sci. 2008, 17, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Röhrborn, D.; Wronkowitz, N.; Eckel, J. DPP4 in diabetes. Front. Immunol. 2015, 6, 386. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.J.; Zhu, M.R.; Lu, C.M.; Jin, Y.; Gao, L.H.; Li, L.; Zhou, J.; Li, F.F.; Zhao, Q.H.; Liu, H.K.; et al. Synthesis and characterization of oxidovanadium complexes as enzyme inhibitors targeting dipeptidyl peptidase IV. J. Inorg. Biochem. 2017, 175, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Meduru, H.; Wang, Y.T.; Tsai, J.; Chen, Y.C. Finding a potential dipeptidyl peptidase-4 (DPP-4) inhibitor for type-2 diabetes treatment based on molecular docking, pharmacophore generation, and molecular dynamics simulation. Int. J. Mol. Sci. 2016, 17, 920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.; Manan, A.; Mirza, M.U. Molecular docking and in silico ADMET studies of silibinin and glycyrrhetic acid anti-inflammatory activity. Trop. J. Pharm. Res. 2017, 16, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Pîrnău, A.; Vlase, L.; Vodnar, D.C.; Duma, M.; Tiperciuc, B.; Oniga, O. 3, 5-Disubstituted thiazolidine-2, 4-diones: Design, microwave-assisted synthesis, antifungal activity, and ADMET screening. SLAS Discov. 2018, 23, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Azad, I.; Nasibullah, M.; Khan, T.; Hassan, F.; Akhter, Y. Exploring the novel heterocyclic derivatives as lead molecules for design and development of potent anticancer agents. J. Mol. Graph. Model. 2018, 81, 211–228. [Google Scholar] [CrossRef]
- Hu, M.; Ling, J.; Lin, H.; Chen, J. Use of Caco-2 cell monolayers to study drug absorption and metabolism. In Optimization in Drug Discovery: In vitro Methods, 1st ed.; Yan, Z., Caldwell, G., Eds.; Humana Press: Totowa, NJ, USA, 2004; Volume 1, pp. 19–35. [Google Scholar]
- Shinde, M.G.; Modi, S.J.; Kulkarni, V.M. Synthesis, pharmacological evaluation, molecular docking and in silico ADMET prediction of nitric oxide releasing biphenyls as anti-inflammatory agents. J. Appl. Pharm. Sci. 2017, 7, 37–47. [Google Scholar]
- Nisha, C.M.; Kumar, A.; Nair, P.; Gupta, N.; Silakari, C.; Tripathi, T.; Kumar, A. Molecular docking and in silico ADMET study reveals acylguanidine 7a as a potential inhibitor of β-secretase. Adv. Bioinform. 2016, 2016, 9258578. [Google Scholar] [CrossRef] [Green Version]
- Encinar, J.A.; Fernández-Ballester, G.; Galiano-Ibarra, V.; Micol, V. In silico approach for the discovery of new PPARγ modulators among plant-derived polyphenols. Drug Des. Dev. Ther. 2015, 9, 5877. [Google Scholar] [CrossRef] [Green Version]
- Iheagwam, F.N.; Nsedu, E.I.; Kayode, K.O.; Emiloju, O.C.; Ogunlana, O.O.; Chinedu, S.N. Bioactive screening and in vitro antioxidant assessment of Nauclea latifolia leaf decoction. AIP Conf. Proc. 2018, 1954, 030015. [Google Scholar]
- Iheagwam, F.N.; Israel, E.N.; Kayode, K.O.; DeCampos, O.C.; Ogunlana, O.O. and Chinedu1, S.N. GCMS analysis and inhibitory evaluation of Terminalia catappa leaf extracts on major enzymes linked to diabetes. Evid. Based Complement. Altern. Med. 2019, 2019, 6316231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Hao, F.; Lai, X.; Yu, H.; Huang, Y.; Wang, H. Expression, purification and properties of shikimate dehydrogenase from MTB. BMB Rep. 2005, 38, 624. [Google Scholar] [CrossRef] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook, 1st ed.; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; Volume 1, pp. 571–607. [Google Scholar]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, P.; Cao, D.S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Compound | Retention Time (min) | Area (%) | Formula | Molecular Weight | Compound Classification |
|---|---|---|---|---|---|---|
| 1 | 2-Furanmethanediol, dipropionate | 5.702 | 0.31 | C11H14O5 | 226 | fatty acid |
| 2 | 2-Furanmethanol | 5.902 | 0.09 | C5H6O2 | 98 | alcohol |
| 3 | 2-Oxopentanedioic acid | 7.45 | 0.09 | C5H6O5 | 146 | carboxylic acid |
| 4 | 1,3-Cyclohexanedione | 7.708 | 0.71 | C6H8O2 | 112 | phenolic |
| 5 | 4-Benzyloxy-6-hydroxymethyl-tetrahydropyran-2,3,5-triol | 8.322 | 0.61 | C13H18O6 | 270 | phenolic |
| 6 | 1H-Azonine, octahydro-1-nitroso- | 9.081 | 0.27 | C8H16N2O | 156 | alkaloid |
| 7 | Phenylethyl alcohol | 9.556 | 0.19 | phenolic | ||
| 8 | 4H-Pyran-4-one,2,3-dihydro-3,5-dihydroxy-6-methyl- # | 10.065 | 0.54 | C6H8O4 | 144 | phenolic |
| 9 | Benzoic acid | 10.476 | 0.41 | C7H6O2 | 122 | phenolic |
| 10 | α-Terpineol *@ | 10.775 | 0.2 | C10H18O | 154 | terpene |
| 11 | 1-(Methoxymethoxy)-3-methyl-3-hydroxybutane | 10.947 | 0.18 | C7H16O3 | 148 | alcohol |
| 12 | Benzofuran, 2,3-dihydro- | 11.083 | 0.53 | C8H8O | 120 | phenolic |
| 13 | 5-Hydroxymethylfurfural | 11.213 | 1.13 | C6H6O3 | 126 | carbohydrate |
| 14 | 2-Methoxy-4-vinylphenol | 12.324 | 0.73 | C9H10O2 | 150 | phenolic |
| 15 | 1,2,4-Benzenetriol * | 13.303 | 0.4 | C6H6O3 | 126 | phenolic |
| 16 | 2-Hydroxy-5-methylisophthalaldehyde | 13.916 | 1.25 | C9H8O3 | 164 | phenolic |
| 17 | 3-Tosylsedoheptulose | 14.434 | 0.12 | C14H20O9S | 364 | carbohydrate |
| 18 | Caprylic anhydride | 14.667 | 0.25 | C16H30O3 | 270 | fatty acid |
| 19 | 4,4-Dimethyl-cyclohex-2-en-1-ol | 14.748 | 0.14 | C8H14O | 126 | phenolic |
| 20 | 5-Caranol, trans, trans-(+)- | 14.868 | 0.14 | C10H18O | 154 | terpenoid |
| 21 | 11-(2-Cyclopenten-1-yl) undecanoicacid, (+)- | 14.981 | 0.11 | C16H28O2 | 252 | fatty acid |
| 22 | 4-Hydroxy-2-hydroxyaminopyrimidine | 15.062 | 0.35 | C4H5N3O2 | 127 | alkaloid |
| 23 | 9-Oxabicyclo[3.3.1]nonane-2,6-diol | 15.143 | 0.77 | C8H14O3 | 158 | phenolic |
| 24 | Megastigmatrienone | 15.627 | 0.23 | C13H18O | 190 | terpene |
| 25 | 2-Cyclohexen-1-one, 4-(3-hydroxy-1-butenyl)-3,5,5-trimethyl-, [R-[R*,R*-(E)]]- | 15.768 | 0.97 | C13H20O2 | 208 | terpene |
| 26 | 1,2,3,5-Cyclohexanetetrol, (1-alpha,2-beta,3-alpha,5-beta)- | 16.278 | 3.64 | C6H12O4 | 148 | phenolic |
| 27 | Tridecanoic acid | 16.592 | 1.38 | C13H26O2 | 214 | fatty acid |
| 28 | Dodecanoic acid * | 16.77 | 2.09 | C12H24O2 | 200 | fatty acid |
| 29 | [1,1’-Bicyclopropyl]-2-octanoic acid, 2’-hexyl-, methyl ester | 16.908 | 2.22 | C21H38O2 | 322 | fatty acid ester |
| 30 | 2-O-p-Methylphenyl-1-thio-β-d-glucoside | 16.984 | 2.47 | C13H18O5S | 286 | carbohydrate |
| 31 | 3-O-Methyl-d-glucose | 17.333 | 2.68 | C7H14O6 | 194 | carbohydrate |
| 32 | n-Hexadecanoic acid #* | 18.126 | 10.86 | C16H32O2 | 256 | fatty acid |
| 33 | Hexadecanoic acid, ethyl ester * | 18.195 | 7.32 | C18H36O2 | 284 | fatty acid ester |
| 34 | γ-Sitosterol *@ | 18.375 | 1.18 | C29H50O | 414 | terpenoid |
| 35 | 9,9-Dimethoxybicyclo[3.3.1]nona-2,4-dione | 18.814 | 1.04 | C11H16O4 | 212 | phenolic |
| 36 | Phytol #@ | 18.983 | 7.24 | C20H40O | 296 | terpenoid |
| 37 | Ethyl Oleate | 19.319 | 18 | C20H38O2 | 310 | fatty acid ester |
| 38 | Octadecanoic acid, ethyl ester # | 19.441 | 5.51 | C20H40O2 | 312 | fatty acid ester |
| 39 | Androstan-17-one, 16,16-dimethyl-(5-alpha)- | 19.824 | 3.05 | C21H34O | 302 | terpenoid |
| 40 | 1-Naphthalenol,decahydro-1,4a-dimethyl-7-(1-methylethylidene)- | 20.088 | 2.69 | C15H26O | 222 | phenolic |
| 41 | 17-Octadecynoic acid | 20.721 | 0.29 | C11H21N | 280 | fatty acid |
| S/N | Compound | Retention Time (min) | Area (%) | Formula | Molecular Weight | Compound Classification |
|---|---|---|---|---|---|---|
| 1 | 2,3-Butanediol | 5.805 | 20.04 | C4H10O2 | 90 | alcohol |
| 2 | 2,5-Dimethyl-4-hydroxy-3(2H)-furanone | 8.869 | 1.32 | C6H8O3 | 128 | phenolic |
| 3 | Catechol * | 10.886 | 14.84 | C6H6O2 | 110 | phenolic |
| 4 | Benzofuran, 2,3-dihydro- | 11.107 | 0.74 | C8H8O | 120 | phenolic |
| 5 | 2-Methoxy-4-vinylphenol | 12.339 | 0.81 | C9H10O2 | 150 | phenolic |
| 6 | 2,7-Octadiene-1,6-diol, 2,6-dimethyl- | 12.914 | 0.38 | C10H18O2 | 170 | terpene |
| 7 | 2-Hydroxy-5-methylisophthalaldehyde | 13.893 | 1.16 | C9H8O3 | 164 | phenolic |
| 8 | Bicyclo[2.2.1]heptan-2-one, 1-(bromomethyl)-7,7-dimethyl-,(1S)- | 14.764 | 0.21 | C10H15BrO | 230 | phenolic |
| 9 | 11-(2-Cyclopenten-1-yl)undecanoic acid, (+)- | 14.982 | 0.9 | C16H28O2 | 252 | fatty acid |
| 10 | 9-Oxabicyclo[3.3.1]nonane-2,6-diol | 15.384 | 19.75 | C8H14O3 | 158 | phenolic |
| 11 | 2-Cyclohexen-1-one, 4-(3-hydroxy-1-butenyl)-3,5,5-trimethyl-, [R-[R*,R*-(E)]]- | 15.807 | 1.67 | C13H20O2 | 208 | terpene |
| 12 | 9,9-Dimethoxybicyclo[3.3.1]nona-2,4-dione | 16.052 | 3.44 | C11H16O4 | 212 | phenolic |
| 13 | 5,5,8a-Trimethyl-3,5,6,7,8,8a-hexahydro-2H-chromene | 17.07 | 11.13 | C12H20O | 180 | phenolic |
| 14 | [1,1’-Bicyclopropyl]-2-octanoic acid, 2’-hexyl-, methyl ester | 17.522 | 12.01 | C21H38O2 | 322 | fatty acid ester |
| 15 | n-Hexadecanoic acid #* | 18.035 | 0.89 | C16H32O2 | 256 | fatty acid |
| 16 | Ethyl 14-methyl-hexadecanoate | 18.171 | 0.23 | C19H38O2 | 298 | fatty acid |
| 17 | Vitamin E #* | 18.481 | 2.89 | C22H30O5 | 430 | terpenoid |
| 18 | Phytol #@ | 18.96 | 0.07 | C20H40O | 296 | terpenoid |
| 19 | 9,12-Octadecadienoic acid (Z,Z)- #@ | 19.217 | 5.03 | C18H32O2 | 280 | fatty acid |
| S/N | Template | GMQE | QSQE | Sequence Identity | Sequence Similarity | Resolution | Oligomeric State |
|---|---|---|---|---|---|---|---|
| 1 | 1wcy | 0.99 | 1 | 100 | 0.62 | 2.2 Å | homo-dimer |
| 2 | 3qbj | 0.99 | 1 | 99.73 | 0.62 | 2.2 Å | homo-dimer |
| 3 | 2qt9 | 0.99 | 1 | 99.87 | 0.62 | 2.1 Å | homo-dimer |
| 4 | 2bgr | 0.99 | 0.96 | 100 | 0.62 | 2.0 Å | homo-dimer |
| 5 | 5lls | 0.94 | 1 | 88.38 | 0.59 | 2.4 Å | homo-dimer |
| 6 | 2gbg | 0.94 | 1 | 84.99 | 0.58 | 3.0 Å | homo-dimer |
| 7 | 2jid | 0.99 | 0.93 | 100 | 0.62 | 2.8 Å | homo-dimer |
| 8 | 1orv | 0.97 | 0.68 | 88.19 | 0.59 | 1.8 Å | homo-tetramer |
| 9 | 3f8s | 0.99 | - | 100 | 0.62 | 2.4 Å | monomer |
| 10 | 5vta | 0.94 | - | 85.01 | 0.58 | 2.8 Å | hetero-trimer |
| 11 | 4ffv | 0.93 | 1 | 84.99 | 0.58 | 2.4 Å | hetero-hexamer |
| Model No | 3D Refine Score | RWplus | MolProbity |
|---|---|---|---|
| 5 | 31,326.3 | −174,719.07 | 1.412 |
| 4 | 31,591.2 | −174,480.02 | 1.302 |
| 3 | 31,993.4 | −174,208.07 | 1.344 |
| 2 | 32,698.8 | −174,067.52 | 1.258 |
| 1 | 35,422.7 | −174,035.11 | 1.190 |
| PROCHECK | G-Factor 2 | ||||||
|---|---|---|---|---|---|---|---|
| Most Favored (%) | Additional Allowed (%) | Generously Allowed (%) | Disallowed (%) | Torsion Angles | Covalent Geometry | Overall Average | |
| DPP4.A | 89.6 | 10.1 | 0.2 | 0.2 | −0.20 | 0.11 | −0.07 |
| Min DPP4 | 88.8 | 10.6 | 0.3 | 0.3 | −0.02 | −1.07 | −0.52 |
| 1wcy.A | 86.7 | 12.9 | 0.3 | 0.2 | 0.13 | 0.54 | 0.30 |
| Docking Score (kcal/mol) | ||||||
|---|---|---|---|---|---|---|
| S/N | Compound | IGEMDOCK | AutoDock Vina | |||
| TE | VdW | HB | Elec | BE | ||
| 1 | 1,2,3,5-Cyclohexanetetrol, 1-alpha,2-beta,3-alpha,5-beta- | −64.14 | −32.47 | −31.67 | 0.00 | −5.2 |
| 2 | 1,2,4-Benzenetriol | −65.06 | −43.51 | −21.54 | 0.00 | −4.9 |
| 3 | 1,3-Cyclohexanedione | −58.68 | −43.60 | −15.08 | 0.00 | −4.3 |
| 4 | 1-(Methoxymethoxy)-3-methyl-3-hydroxybutane | −67.65 | −51.15 | −16.50 | 0.00 | −4.3 |
| 5 | 1-Naphthalenol,decahydro-1,4a-dimethyl-7-(1-methylethylidene) | −64.18 | −58.18 | −6.00 | 0.00 | −6.5 |
| 6 | 11-(2-Cyclopenten-1-yl)undecanoicacid, (+)- | −70.60 | −63.76 | −8.77 | 1.93 | −5.1 |
| 7 | 17-Octadecynoic acid | −73.26 | −63.31 | −9.89 | −0.06 | −4.9 |
| 8 | 1H-Azonine, octahydro-1-nitroso- | −54.18 | −41.95 | −12.23 | 0.00 | −4.7 |
| 9 | 2,3-Butanediol | −51.07 | −36.18 | −14.89 | 0.00 | −4.3 |
| 10 | 2,5-Dimethyl-4-hydroxy-3(2H)-furanone | −56.19 | −44.76 | −11.43 | 0.00 | −4.8 |
| 11 | 2,7-Octadiene-1,6-diol, 2,6-dimethyl- | −62.21 | −52.88 | −9.33 | 0.00 | −4.8 |
| 12 | 2-Cyclohexen-1-one, 4-(3-hydroxy-1-butenyl)-3,5,5-trimethyl-, [R-[R*,R*-(E)]]- | −61.02 | −55.77 | −5.25 | 0.00 | −6.1 |
| 13 | 2-Furanmethanediol, dipropionate | −68.62 | −59.32 | −9.30 | 0.00 | −5.7 |
| 14 | 2-Furanmethanol | −56.91 | −37.00 | −19.91 | 0.00 | −4.4 |
| 15 | 2-Hydroxy-5-methylisophthalaldehyde | −71.18 | −55.30 | −15.88 | 0.00 | −5.1 |
| 16 | 2-Methoxy-4-vinylphenol | −70.07 | −54.15 | −15.92 | 0.00 | −5.1 |
| 17 | 2-O-p-Methylphenyl-1-thio-β-d-glucoside | −76.67 | −60.32 | −16.35 | 0.00 | −6.9 |
| 18 | 2-Oxopentanedioic acid | −64.36 | −39.96 | −20.20 | −4.20 | −5.2 |
| 19 | 3-O-Methyl-d-Glucose | −79.56 | −48.45 | −31.12 | 0.00 | −5 |
| 20 | 3-Tosylsedoheptulose | −79.45 | −64.19 | −15.26 | 0.00 | −7 |
| 21 | 4,4-Dimethyl-cyclohex-2-en-1-ol | −51.95 | −39.09 | −12.87 | 0.00 | −4.6 |
| 22 | 4-Benzyloxy-6-hydroxymethyl-tetrahydropyran-2,3,5-triol | −90.22 | −59.82 | −30.40 | 0.00 | −6.8 |
| 23 | 4-Hydroxy-2-hydroxyaminopyrimidine | −72.29 | −47.82 | −24.47 | 0.00 | −5.5 |
| 24 | 4H-Pyran-4-one,2,3-dihydro-3,5-dihydroxy-6-methyl- | −73.41 | −44.81 | −28.61 | 0.00 | −5.3 |
| 25 | 5,5,8a-Trimethyl-3,5,6,7,8,8a-hexahydro-2H-chromene | −59.09 | −55.59 | −3.50 | 0.00 | −5.6 |
| 26 | 5-Caranol, trans, trans-(+)- | −57.52 | −44.52 | −13.00 | 0.00 | −5.1 |
| 27 | 5-Hydroxymethylfurfural | −72.97 | −57.68 | −15.29 | 0.00 | −5.1 |
| 28 | 9,12-Octadecadienoic acid (Z,Z)- | −72.42 | −68.72 | −2.34 | −1.37 | −5.2 |
| 29 | 9,9-Dimethoxybicyclo[3.3.1]nona-2,4-dione | −66.88 | −52.88 | −14.00 | 0.00 | −4.8 |
| 30 | 9-Oxabicyclo[3.3.1]nonane-2,6-diol | −69.24 | −49.12 | −20.12 | 0.00 | −4.8 |
| 31 | [1,1’-Bicyclopropyl]-2-octanoic acid, 2’-hexyl-, methyl ester | −70.68 | −67.18 | −3.50 | 0.00 | −6 |
| 32 | α-Terpineol | −63.97 | −58.97 | −5.00 | 0.00 | −5.2 |
| 33 | Androstan-17-one, 16, 16-dimethyl-(5.alpha.)- | −63.14 | −60.74 | −2.40 | 0.00 | −7.3 |
| 34 | Benzofuran, 2,3-dihydro- | −54.13 | −44.07 | −10.06 | 0.00 | −4.7 |
| 35 | Benzoic acid | −56.65 | −47.63 | −9.23 | 0.21 | −5 |
| 36 | Bicyclo[2.2.1]heptan-2-one, 1-(bromomethyl)-7,7-dimethyl-,(1S)- | −56.79 | −46.29 | −10.50 | 0.00 | |
| 37 | Caprylic anhydride | −65.63 | −56.09 | −9.54 | 0.00 | −4.6 |
| 38 | Cathecol | −65.43 | −43.47 | −21.95 | 0.00 | −4.4 |
| 39 | Dodecanoicacid | −50.00 | −45.96 | −4.04 | 0.00 | −4.6 |
| 40 | Ethyl 14-methyl-hexadecanoate | −66.40 | −62.90 | −3.50 | 0.00 | −5.1 |
| 41 | Ethyl Oleate | −74.10 | −69.11 | −4.99 | 0.00 | −5.4 |
| 42 | Hexadecanoic acid, ethyl ester | −63.14 | −59.64 | −3.50 | 0.00 | −4.5 |
| 43 | n-Hexadecanoic acid | −71.33 | −59.08 | −10.50 | −1.75 | −4.4 |
| 44 | Megastigmatrienone | −62.48 | −58.82 | −3.66 | 0.00 | −6.2 |
| 45 | Octadecanoic acid, ethyl ester | −64.67 | −61.23 | −3.44 | 0.00 | −4.9 |
| 46 | Phenylethyl Alcohol | −61.00 | −47.87 | −13.14 | 0.00 | −4.7 |
| 47 | Phytol | −70.24 | −66.74 | −3.50 | 0.00 | −5.6 |
| 48 | Tridecanoic Acid | −60.93 | −58.06 | −3.50 | 0.62 | −4.5 |
| 49 | Vitamin E | −77.10 | −67.80 | −9.30 | 0.00 | −6.7 |
| 50 | γ-Sitosterol | −69.45 | −65.95 | −3.50 | 0.00 | −7.8 |
| 51 | Alogliptin | −82.55 | −63.39 | −19.17 | 0.00 | −6.7 |
| 52 | Saxagliptin | −76.74 | −51.85 | −24.90 | 0.00 | −6.7 |
| Compound | Hb | CHb | VdW | π-π |
|---|---|---|---|---|
| 2-O-p-Methylphenyl-1-thio-β-d-glucoside | Lys122, Asp709, Ala707, Tyr238 | Gln123 | Val252, Ser744, Trp124, Asp737, Glu738 | Asp739, Phe240, Ala743 |
| 3-Tosylsedoheptulose | Tyr381, Gln 586, Lys 598, Ile 590 | Lys523 | Thr401, Trp402, Glu403, Asn420, Glu521, Thr522, Trp525, Tyr585, Gly587 | Pro426 |
| 4-Benzyloxy-6-hydroxymethyl-tetrahydropyran-2,3,5-triol | Lys122, Gly741, Asp739, His740 | Arg125 | Gln123, Trp124, Ser630, Asp709, Asn710, Ala743 | Trp629, His740 |
| Vitamin E | Lys392, Asp393, Cys394 | Thr350, Thr351, Ser349, Ser376, Glu378, Glu347, His592, Asp588, Phe387, Asn377, Met348, Ile346 | Ile375, Cys385, 394, Phe396, Lys392 | |
| Saxagliptin | Lys175, Ile148, Asn150, Ser182 | Tyr166, Glu146, Pro149, 181, | Arg147 | |
| Alogliptin | His740, Trp629, Lys554 | Ser630 | Gly628, Gly632, Gly741, Val546, Trp627, Tyr752 | Trp629, Tyr547 |
| Compound | Hb | CHb | VdW | π-π |
|---|---|---|---|---|
| 2-O-p-Methylphenyl-1-thio-β-d-glucoside | Tyr547, Tyr662, Glu205, Glu206, Asn710, Ser630, His740 | Arg125, Arg358, Ser209, Trp629, Val711, Tyr666 | Phe357 | |
| 3-Tosylsedoheptulose | Glu206, Tyr662, Tyr547, Ser630, Arg125, His740 | Arg669, Phe357, Tyr666, Tyr631, Lys554, Trp629, Glu205 | His740, Tyr547 | |
| 4-Benzyloxy-6-hydroxymethyl-tetrahydropyran-2,3,5-triol | Arg125, Arg358, Ser209, Glu206 | Glu205 | Pro550, Ser552, Gly549, Arg669, Tyr666, | Tyr547, Phe357 |
| Vitamin E | Glu206 | His740, Trp629, Ser209, Ser552, Ser630, Gln553, Arg358, Arg669, Glu205, | Phe357, Tyr547, Tyr666, Lys554 | |
| Saxagliptin | Ser209, Ser630, Arg125, Tyr547 | Tyr547 | Glu206, Glu205, Tyr631, Tyr662, His126, His740, Arg358 | Phe357, Tyr666 |
| Alogliptin | Arg125, Arg358, Glu205, Glu206, Asn710 | Ser209, Arg358 | Tyr662 | Phe357, Tyr 666 |
| 2-O-p-Methylphenyl-1-thio-β-d-glucoside | 3-Tosyl sedoheptulose | 4-Benzyloxy-6-hydroxymethyl-tetrahydropyran-2,3,5-triol | Vitamin E | Lipinski Rule Details | |
|---|---|---|---|---|---|
| MW (g/mol) | 286.34 | 364.37 | 270.281 | 430.71 | ≤ 500 |
| Hb donor | 4 | 5 | 4 | 1 | ≤ 5 |
| Hb acceptor | 5 | 9 | 6 | 2 | ≤ 10 |
| LogP | −0.113 | −2.137 | −0.997 | 8.84 | ≤ 5 |
| TPSA | 115.45 | 162.13 | 99.38 | 29.46 | - |
| NRb | 3 | 5 | 4 | 12 | - |
| MR | 70.78 | 79.25 | 64.95 | 139.27 | - |
| # Atoms | 37 | 44 | 37 | 81 | - |
| # Lipinski Violations | - | - | - | 1 |
| 2-O-p-Methylphenyl-1-thio-β-D-glucoside | 3-Tosyl sedoheptulose | 4-Benzyloxy-6-hydroxymethyl-tetrahydropyran-2,3,5-triol | Vitamin E | |
|---|---|---|---|---|
| Absorption | ||||
| Caco-2 permeability (cm/s) | − (−5.697) | − (−6.137) | − (−5.745) | + (−4.969) |
| Blood Brain Barrier | ++ | − | ++ | +++ |
| Human Intestinal Absorption | −−− | −−− | −−− | ++ |
| P-glycoprotein Inhibitor | −−− | − | −−− | − |
| P-glycoprotein Substrate | −−− | −−− | −−− | −−− |
| F (20% Bioavailability) | + | − | − | − |
| F (30% Bioavailability) | − | − | − | − |
| Renal Organic Cation Transporter | − | − | − | − |
| Distribution | ||||
| Subcellular localization | Mitochondria | Lysosome | Mitochondria | Mitochondria |
| Plasma Protein Binding (%) | 55.94 | 49.26 | 43.47 | 84.65 |
| Volume Distribution (L/kg) | −0.496 | −1.017 | −0.278 | 0.444 |
| Metabolism | ||||
| P450 CYP1A2 inhibitor | −−− | −−− | −−− | −−− |
| P450 CYP1A2 Substrate | −−− | −−− | −−− | − |
| P450 CYP3A4 inhibitor | −−− | −−− | −−− | − |
| P450 CYP3A4 substrate | − | − | −−− | ++ |
| P450 CYP2C9 inhibitor | −−− | −−− | −−− | −−− |
| P450 CYP2C9 substrate | − | + | − | + |
| P450 CYP2C19 inhibitor | − | − | −−− | −−− |
| P450 CYP2C19 substrate | − | − | − | + |
| P450 CYP2D6 inhibitor | −−− | −−− | − | −−− |
| P450 CYP2D6 substrate | − | − | − | − |
| CYP Inhibitory Promiscuity | Low | Low | Low | Low |
| Excretion | ||||
| T1/2 (h) | 0.98 | 1.21 | 0.93 | 1.95 |
| Clearance Rate (mL/min/kg) | 1.550 | 0.724 | 1.732 | 1.581 |
| Toxicity | ||||
| Human Ether-a-go-go-Related Gene Blockers | − | − | − | − |
| AMES Mutagenicity | −−− | −−− | −−− | −−− |
| Skin sensitization | − | − | −−− | − |
| LD50 (mg/kg) | 736.03 | 1146.95 | 1967.05 | 1161.96 |
| FDA Maximum Recommended Daily Dose | ++ | ++ | ++ | + |
| Carcinogens | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen |
| Acute Oral Toxicity | III | III | III | III |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iheagwam, F.N.; Ogunlana, O.O.; Chinedu, S.N. Model Optimization and In Silico Analysis of Potential Dipeptidyl Peptidase IV Antagonists from GC-MS Identified Compounds in Nauclea latifolia Leaf Extracts. Int. J. Mol. Sci. 2019, 20, 5913. https://doi.org/10.3390/ijms20235913
Iheagwam FN, Ogunlana OO, Chinedu SN. Model Optimization and In Silico Analysis of Potential Dipeptidyl Peptidase IV Antagonists from GC-MS Identified Compounds in Nauclea latifolia Leaf Extracts. International Journal of Molecular Sciences. 2019; 20(23):5913. https://doi.org/10.3390/ijms20235913
Chicago/Turabian StyleIheagwam, Franklyn Nonso, Olubanke Olujoke Ogunlana, and Shalom Nwodo Chinedu. 2019. "Model Optimization and In Silico Analysis of Potential Dipeptidyl Peptidase IV Antagonists from GC-MS Identified Compounds in Nauclea latifolia Leaf Extracts" International Journal of Molecular Sciences 20, no. 23: 5913. https://doi.org/10.3390/ijms20235913
APA StyleIheagwam, F. N., Ogunlana, O. O., & Chinedu, S. N. (2019). Model Optimization and In Silico Analysis of Potential Dipeptidyl Peptidase IV Antagonists from GC-MS Identified Compounds in Nauclea latifolia Leaf Extracts. International Journal of Molecular Sciences, 20(23), 5913. https://doi.org/10.3390/ijms20235913