Rhomboid-Like-2 Intramembrane Protease Mediates Metalloprotease-Independent Regulation of Cadherins

 , , ,
, , ,  and
and 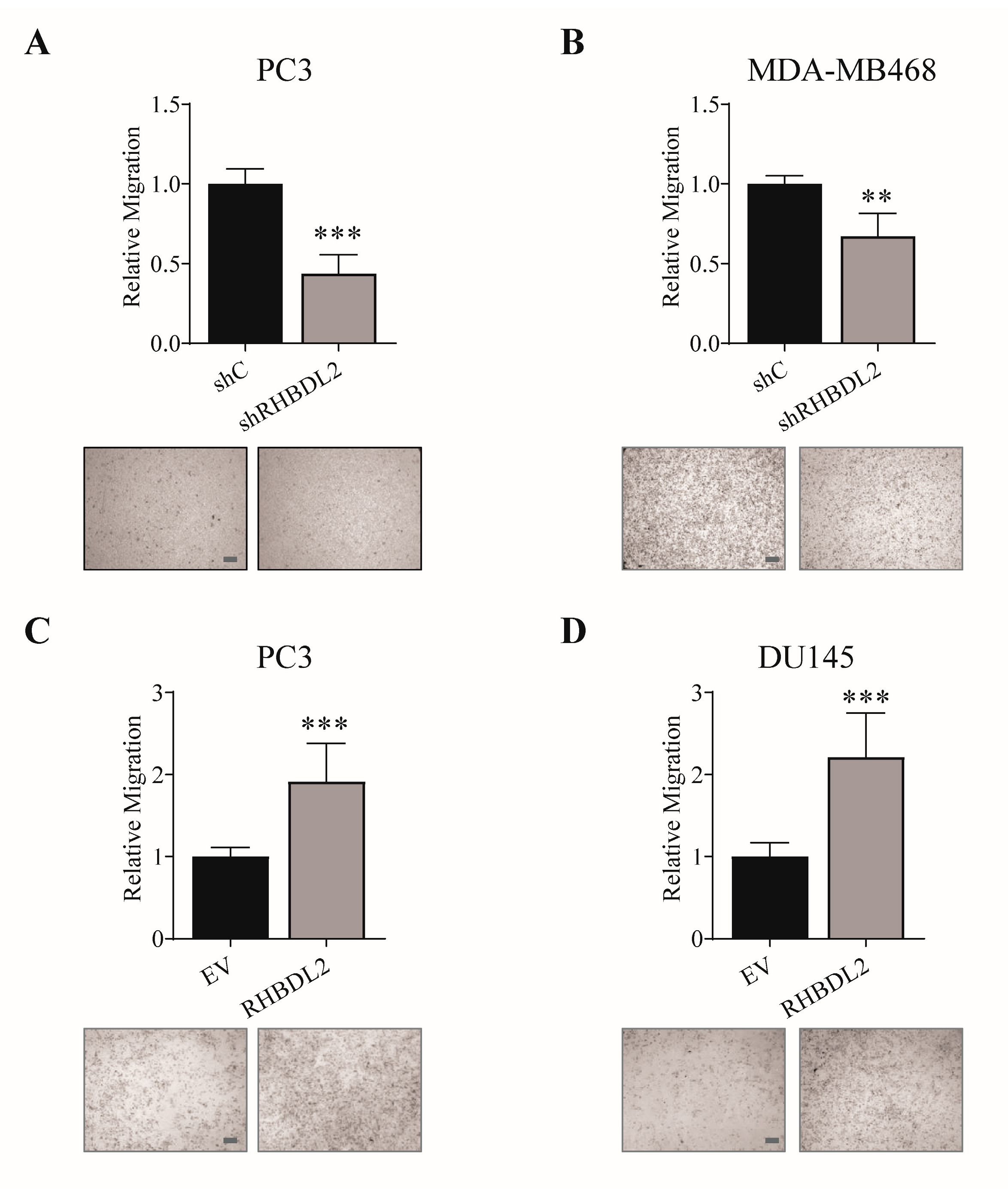{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. RHBDL2 Controls Cancer Cell Migration
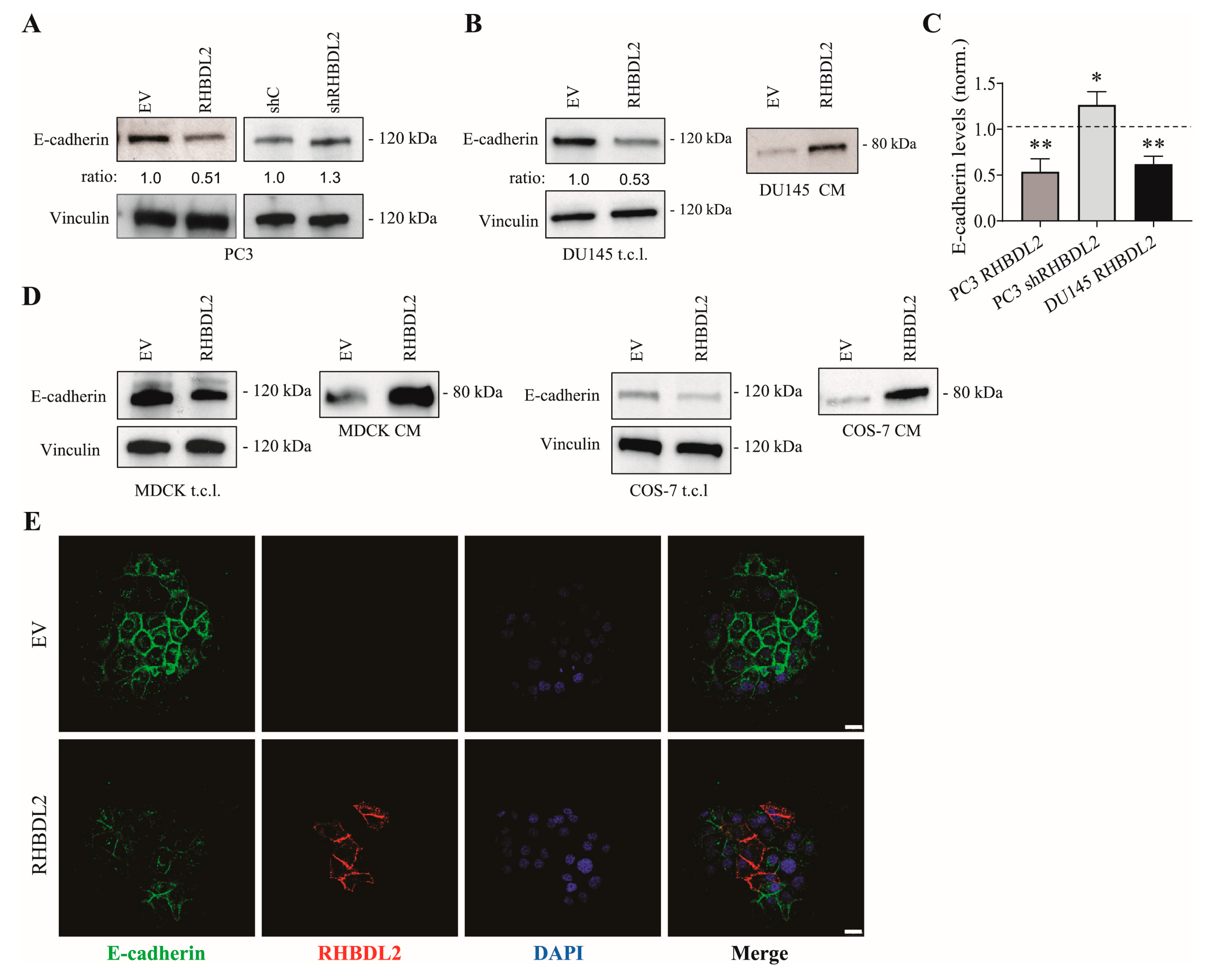
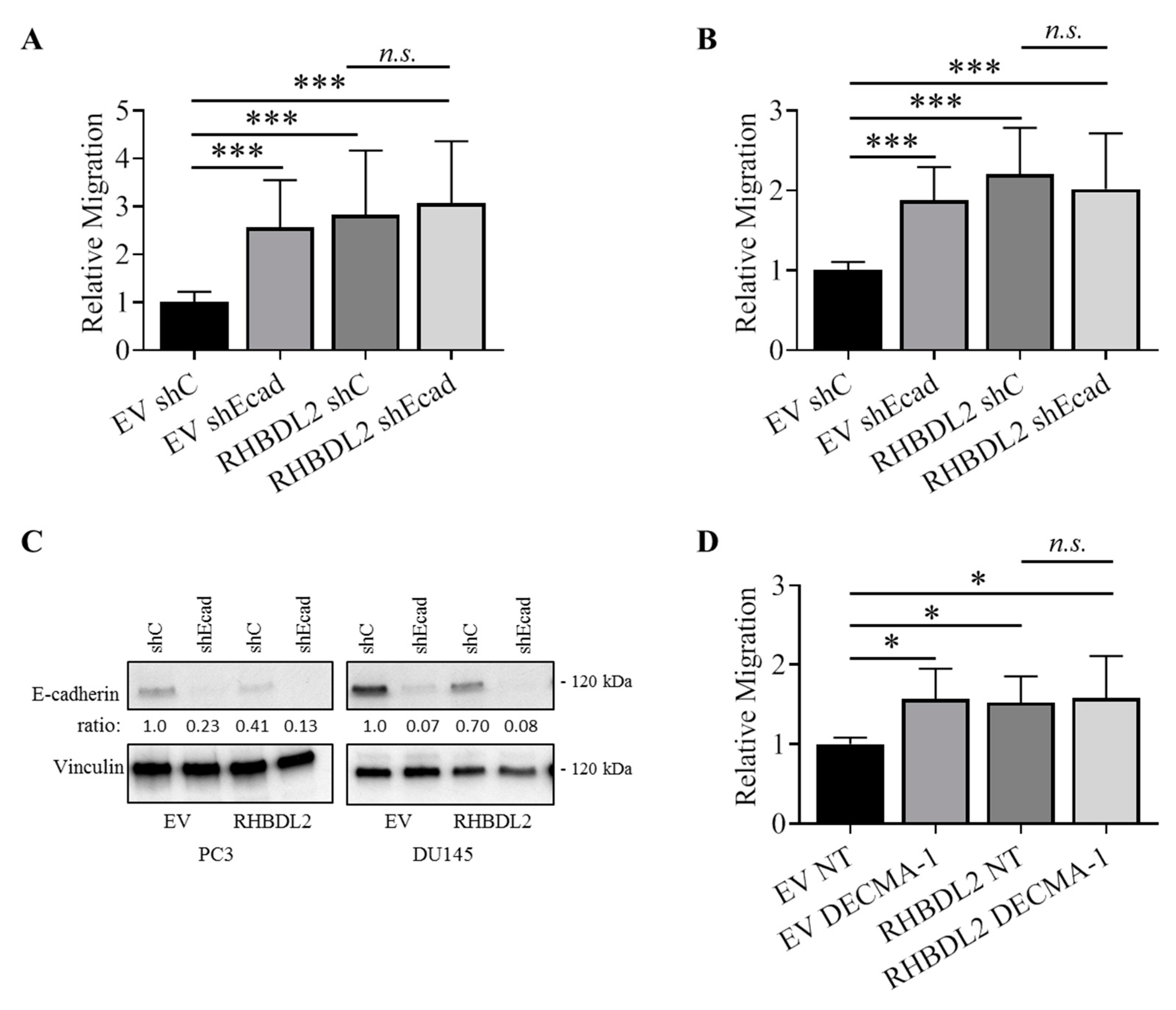
2.2. RHBDL2 Modulates E-Cadherin Expression and Shedding in Human Cancer Cells
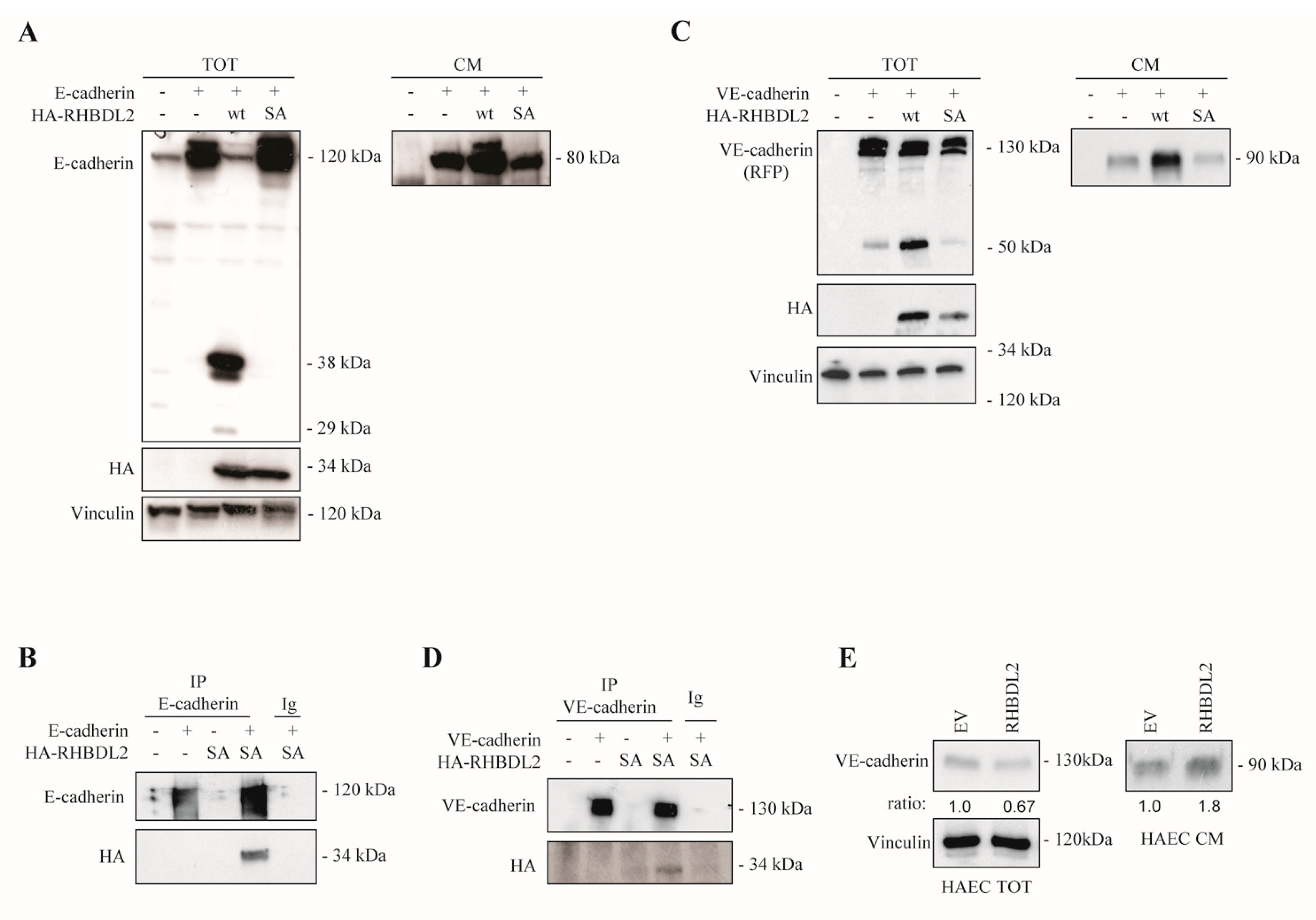
2.3. RHBDL2 Interacts with E-Cadherin and VE-Cadherin, and Catalyzes Their Cleavage and Extracellular Shedding
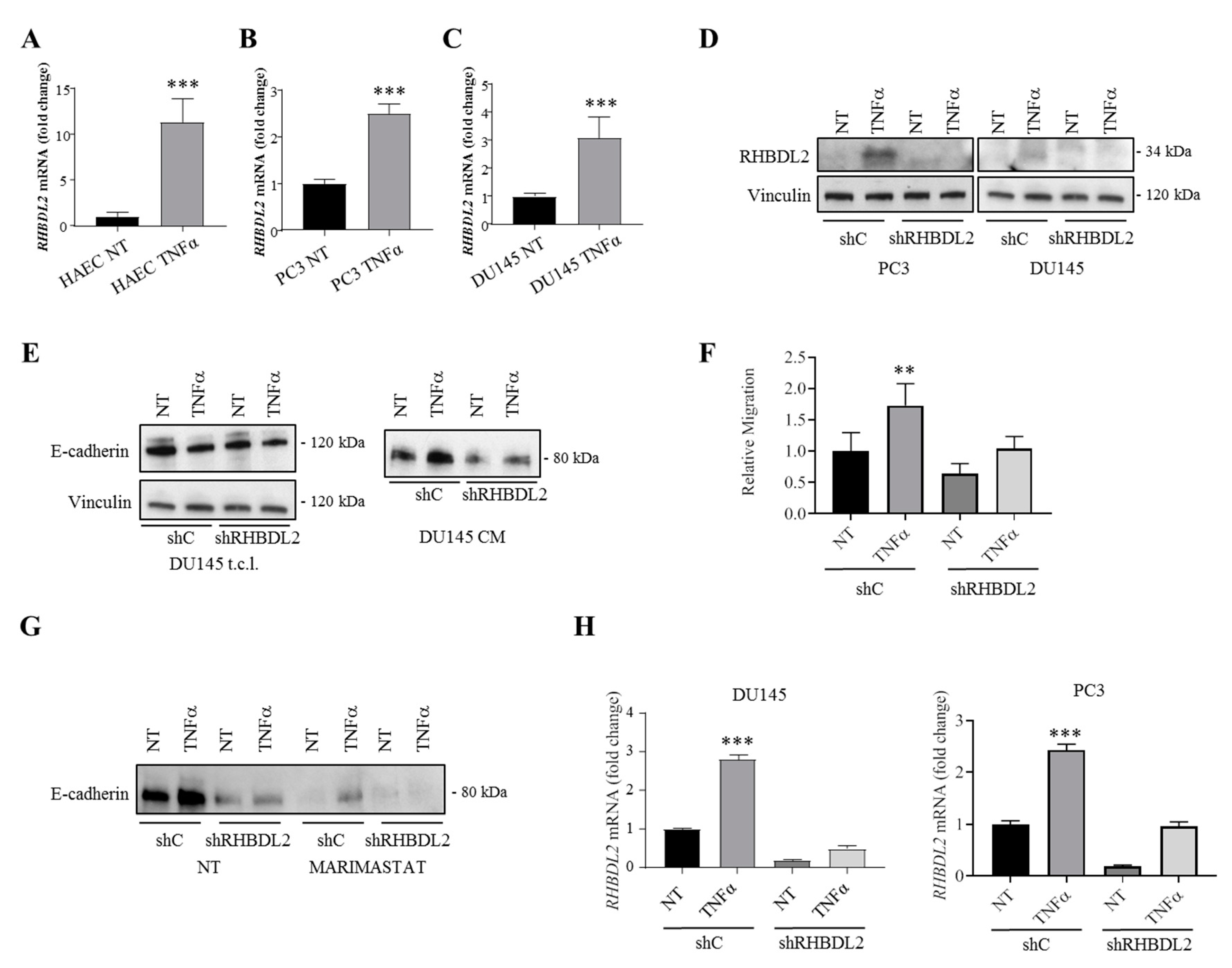
2.4. RHBDL2 Expression Is Induced by the Inflammatory Cytokine TNFα
2.5. RHBDL2 Mediates MMP-Independent E-Cadherin Cleavage in Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Antibodies and Chemicals
4.3. Cell Proliferation Analysis
4.4. Transwell Migration Assays
4.5. Haptotactic Migration Assays with xCELLigence System
4.6. Wound Healing Migration Assays
4.7. Lentiviral-Mediated shRNA or Gene Transfer
4.8. RNA Isolation and Real-Time Quantitative PCR
4.9. Transient Transfection
4.10. Immunoprecipitation and Western Blotting Analysis
4.11. Immunofluorescence
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RHBDL2 | Rhomboid-like-2 |
| MMP | Matrix Metalloprotease |
| TNFα | Tumor Necrosis Factor α |
| EGF | Epidermal Growth Factor |
| ADAM | A Disintegrin And Metalloproteinase |
References
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Fearon, E.R. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer 2002, 34, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5, e1356900. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef]
- Pieters, T.; van Roy, F. Role of cell-cell adhesion complexes in embryonic stem cell biology. J. Cell Sci. 2014, 127, 2603–2613. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Wong, A.S.; Gumbiner, B.M. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J. Cell Biol. 2003, 161, 1191–1203. [Google Scholar] [CrossRef]
- Chunthapong, J.; Seftor, E.A.; Khalkhali-Ellis, Z.; Seftor, R.E.; Amir, S.; Lubaroff, D.M.; Heidger, P.M.; Hendrix, M.J. Dual roles of E-cadherin in prostate cancer invasion. J. Cell Biochem. 2004, 91, 649–661. [Google Scholar] [CrossRef]
- Mao, Q.; Zheng, X.; Yang, K.; Qin, J.; Bai, Y.; Jia, X.; Li, Y.; Xie, L. Suppression of migration and invasion of PC3 prostate cancer cell line via activating E-cadherin expression by small activating RNA. Cancer Investig. 2010, 28, 1013–1018. [Google Scholar] [CrossRef]
- Brown, M.S.; Ye, J.; Rawson, R.B.; Goldstein, J.L. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 2000, 100, 391–398. [Google Scholar] [CrossRef]
- Weihofen, A.; Martoglio, B. Intramembrane-cleaving proteases: Controlled liberation of proteins and bioactive peptides. Trends Cell Biol. 2003, 13, 71–78. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Rogozin, I.B.; Davidovic, L.; Letellier, M.C.; Pellegrini, L. The rhomboids: A nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003, 4, R19. [Google Scholar] [CrossRef]
- Lee, J.R.; Urban, S.; Garvey, C.F.; Freeman, M. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 2001, 107, 161–171. [Google Scholar] [CrossRef]
- Urban, S.; Lee, J.R.; Freeman, M. A family of Rhomboid intramembrane proteases activates all Drosophila membrane-tethered EGF ligands. EMBO J. 2002, 21, 4277–4286. [Google Scholar] [CrossRef]
- Düsterhöft, S.; Künzel, U.; Freeman, M. Rhomboid proteases in human disease: Mechanisms and future prospects. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2200–2209. [Google Scholar] [CrossRef]
- Bergbold, N.; Lemberg, M.K. Emerging role of rhomboid family proteins in mammalian biology and disease. Biochim. Biophys. Acta 2013, 1828, 2840–2848. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Brooke, M.A.; Kelsell, D.P.; Blaydon, D.C. Rhomboid proteins: A role in keratinocyte proliferation and cancer. Cell Tissue Res. 2013, 351, 301–307. [Google Scholar] [CrossRef]
- Verhelst, S.H.L. Intramembrane proteases as drug targets. FEBS J. 2017, 284, 1489–1502. [Google Scholar] [CrossRef]
- Fleig, L.; Bergbold, N.; Sahasrabudhe, P.; Geiger, B.; Kaltak, L.; Lemberg, M.K. Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol. Cell 2012, 47, 558–569. [Google Scholar] [CrossRef]
- Song, W.; Liu, W.; Zhao, H.; Li, S.; Guan, X.; Ying, J.; Zhang, Y.; Miao, F.; Zhang, M.; Ren, X.; et al. Rhomboid domain containing 1 promotes colorectal cancer growth through activation of the EGFR signalling pathway. Nat. Commun. 2015, 6, 8022. [Google Scholar] [CrossRef]
- Wunderle, L.; Knopf, J.D.; Kühnle, N.; Morlé, A.; Hehn, B.; Adrain, C.; Strisovsky, K.; Freeman, M.; Lemberg, M.K. Rhomboid intramembrane protease RHBDL4 triggers ER-export and non-canonical secretion of membrane-anchored TGFα. Sci. Rep. 2016, 6, 27342. [Google Scholar] [CrossRef]
- Pascall, J.C.; Brown, K.D. Intramembrane cleavage of ephrinB3 by the human rhomboid family protease, RHBDL2. Biochem. Biophys. Res. Commun. 2004, 317, 244–252. [Google Scholar] [CrossRef]
- Adrain, C.; Strisovsky, K.; Zettl, M.; Hu, L.; Lemberg, M.K.; Freeman, M. Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep. 2011, 12, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.L.; Wu, Y.T.; Lin, H.Y.; Hsu, F.C.; Liu, S.K.; Chang, B.I.; Chen, W.S.; Lai, C.H.; Shi, G.Y.; Wu, H.L. Functions of rhomboid family protease RHBDL2 and thrombomodulin in wound healing. J. Investig. Dermatol. 2011, 131, 2486–2494. [Google Scholar] [CrossRef]
- Lohi, O.; Urban, S.; Freeman, M. Diverse substrate recognition mechanisms for rhomboids; thrombomodulin is cleaved by Mammalian rhomboids. Curr. Biol. 2004, 14, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Noy, P.J.; Swain, R.K.; Khan, K.; Lodhia, P.; Bicknell, R. Sprouting angiogenesis is regulated by shedding of the C-type lectin family 14, member A (CLEC14A) ectodomain, catalyzed by rhomboid-like 2 protein (RHBDL2). FASEB J. 2016, 30, 2311–2323. [Google Scholar] [CrossRef]
- Menschikowski, M.; Hagelgans, A.; Eisenhofer, G.; Tiebel, O.; Siegert, G. Reducing agents induce thrombomodulin shedding in human endothelial cells. Thromb. Res. 2010, 126, e88–e93. [Google Scholar] [CrossRef]
- Atapattu, L.; Lackmann, M.; Janes, P.W. The role of proteases in regulating Eph/ephrin signaling. Cell Adh. Migr. 2014, 8, 294–307. [Google Scholar] [CrossRef]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef]
- Lastun, V.L.; Grieve, A.G.; Freeman, M. Substrates and physiological functions of secretase rhomboid proteases. Semin. Cell Dev. Biol. 2016, 60, 10–18. [Google Scholar] [CrossRef]
- Ha, Y.; Akiyama, Y.; Xue, Y. Structure and mechanism of rhomboid protease. J. Biol. Chem. 2013, 288, 15430–15436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Baker, R.P.; Ji, M.; Urban, S. Ten catalytic snapshots of rhomboid intramembrane proteolysis from gate opening to peptide release. Nat. Struct. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Březinová, J.; Stephens, E.; Burbridge, E.; Freeman, M.; Adrain, C.; Strisovsky, K. Quantitative proteomics screen identifies a substrate repertoire of rhomboid protease RHBDL2 in human cells and implicates it in epithelial homeostasis. Sci. Rep. 2017, 7, 7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.P.; Tuan, T.L.; Wu, H.; Hughes, M.; Garner, W.L. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J. Cell Sci. 2001, 114, 131–139. [Google Scholar] [PubMed]
- Nee, L.E.; McMorrow, T.; Campbell, E.; Slattery, C.; Ryan, M.P. TNF-alpha and IL-1beta-mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int. 2004, 66, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Hozumi, A.; Nishimura, Y.; Nishiuma, T.; Kotani, Y.; Yokoyama, M. Induction of MMP-9 in normal human bronchial epithelial cells by TNF-alpha via NF-kappa B-mediated pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L1444–L1452. [Google Scholar] [CrossRef]
- Han, Y.P.; Tuan, T.L.; Hughes, M.; Wu, H.; Garner, W.L. Transforming growth factor-beta- and tumor necrosis factor-alpha-mediated induction and proteolytic activation of MMP-9 in human skin. J. Biol. Chem. 2001, 276, 22341–22350. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Noë, V.; Fingleton, B.; Jacobs, K.; Crawford, H.C.; Vermeulen, S.; Steelant, W.; Bruyneel, E.; Matrisian, L.M.; Mareel, M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 2001, 114, 111–118. [Google Scholar]
- Symowicz, J.; Adley, B.P.; Gleason, K.J.; Johnson, J.J.; Ghosh, S.; Fishman, D.A.; Hudson, L.G.; Stack, M.S. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 2007, 67, 2030–2039. [Google Scholar] [CrossRef]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiki, M. The cell surface: The stage for matrix metalloproteinase regulation of migration. Curr. Opin. Cell Biol. 2002, 14, 624–632. [Google Scholar] [CrossRef]
- Cheng, T.L.; Lai, C.H.; Jiang, S.J.; Hung, J.H.; Liu, S.K.; Chang, B.I.; Shi, G.Y.; Wu, H.L. RHBDL2 is a critical membrane protease for anoikis resistance in human malignant epithelial cells. Sci. World J. 2014, 2014, 902987. [Google Scholar] [CrossRef] [PubMed]
- Canzoneri, R.; Lacunza, E.; Isla Larrain, M.; Croce, M.V.; Abba, M.C. Rhomboid family gene expression profiling in breast normal tissue and tumor samples. Tumor Biol. 2014, 35, 1451–1458. [Google Scholar] [CrossRef]
- Chen, H.; Paradies, N.E.; Fedor-Chaiken, M.; Brackenbury, R. E-cadherin mediates adhesion and suppresses cell motility via distinct mechanisms. J. Cell Sci. 1997, 110, 345–356. [Google Scholar]
- Shamir, E.R.; Ewald, A.J. Adhesion in mammary development: Novel roles for E-cadherin in individual and collective cell migration. Curr. Top. Dev. Biol. 2015, 112, 353–382. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.; Cheng, J.C.; Klausen, C.; Leung, P.C.K. Activin A promotes ovarian cancer cell migration by suppressing E-cadherin expression. Exp. Cell Res. 2019, 382, 111471. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Follenzi, A.; Naldini, L. HIV-based vectors. Preparation and use. Methods Mol. Med. 2002, 69, 259–274. [Google Scholar] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battistini, C.; Rehman, M.; Avolio, M.; Arduin, A.; Valdembri, D.; Serini, G.; Tamagnone, L. Rhomboid-Like-2 Intramembrane Protease Mediates Metalloprotease-Independent Regulation of Cadherins. Int. J. Mol. Sci. 2019, 20, 5958. https://doi.org/10.3390/ijms20235958
Battistini C, Rehman M, Avolio M, Arduin A, Valdembri D, Serini G, Tamagnone L. Rhomboid-Like-2 Intramembrane Protease Mediates Metalloprotease-Independent Regulation of Cadherins. International Journal of Molecular Sciences. 2019; 20(23):5958. https://doi.org/10.3390/ijms20235958
Chicago/Turabian StyleBattistini, Chiara, Michael Rehman, Marco Avolio, Alessia Arduin, Donatella Valdembri, Guido Serini, and Luca Tamagnone. 2019. "Rhomboid-Like-2 Intramembrane Protease Mediates Metalloprotease-Independent Regulation of Cadherins" International Journal of Molecular Sciences 20, no. 23: 5958. https://doi.org/10.3390/ijms20235958
APA StyleBattistini, C., Rehman, M., Avolio, M., Arduin, A., Valdembri, D., Serini, G., & Tamagnone, L. (2019). Rhomboid-Like-2 Intramembrane Protease Mediates Metalloprotease-Independent Regulation of Cadherins. International Journal of Molecular Sciences, 20(23), 5958. https://doi.org/10.3390/ijms20235958