MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1β in Renal Cell Carcinoma through ER Stress-Mediated Apoptosis

 ,
,  and
and
Abstract
:1. Introduction
2. Results
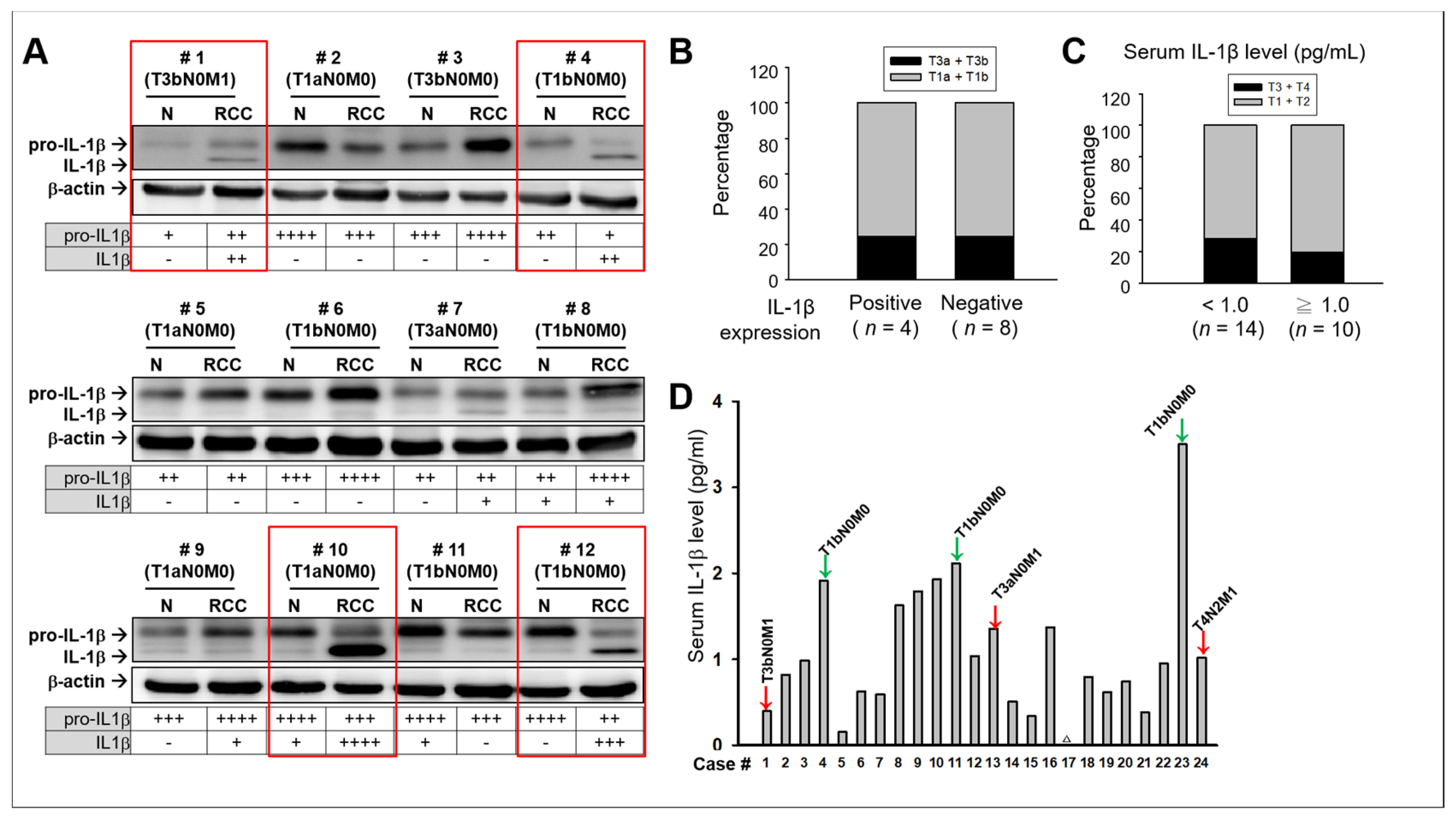
2.1. No Significant Association Between IL-1β and Clinical Consequences in Patients with RCC
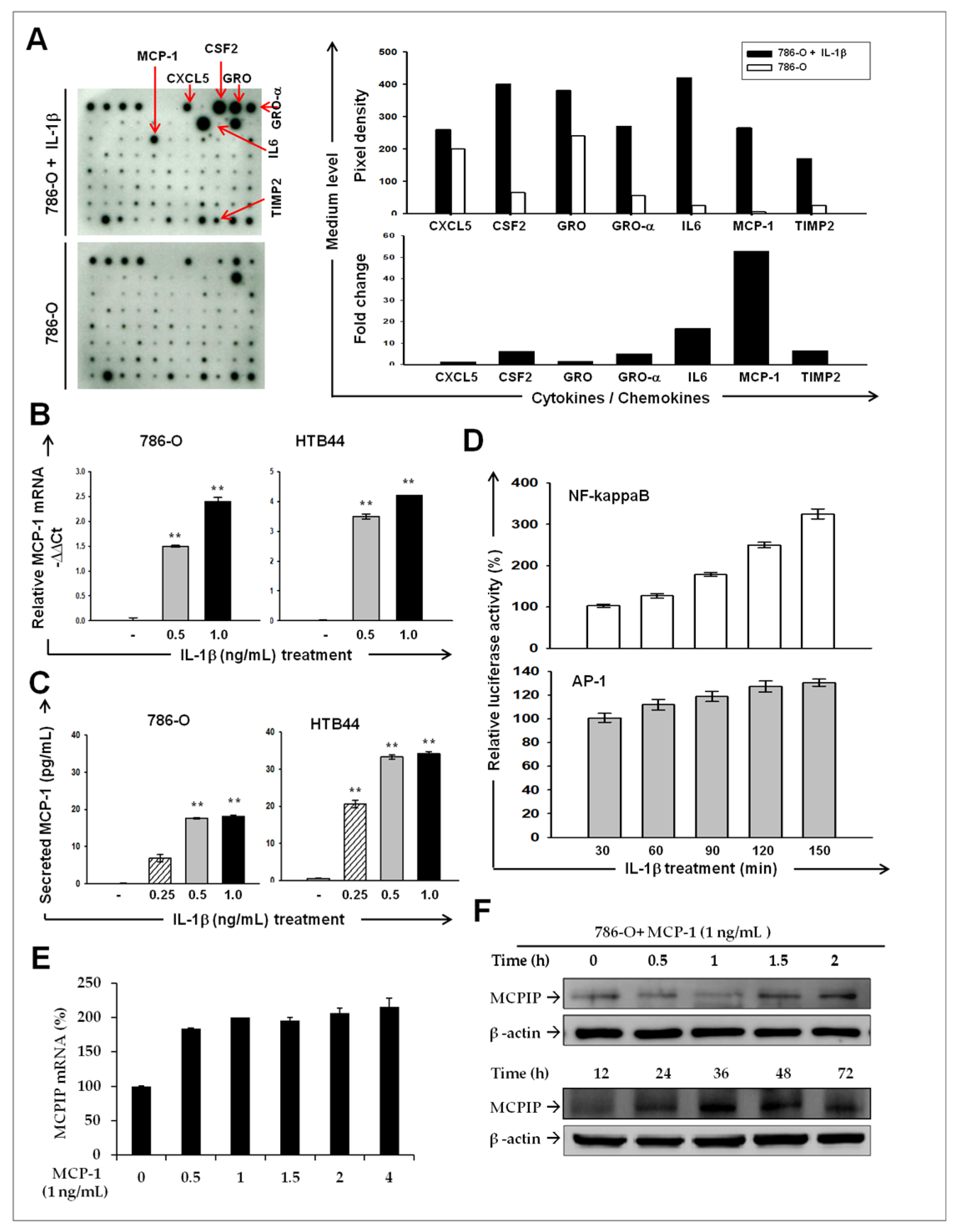
2.2. IL-1β Induces MCP-1/ MCPIP-1 Signaling in RCC
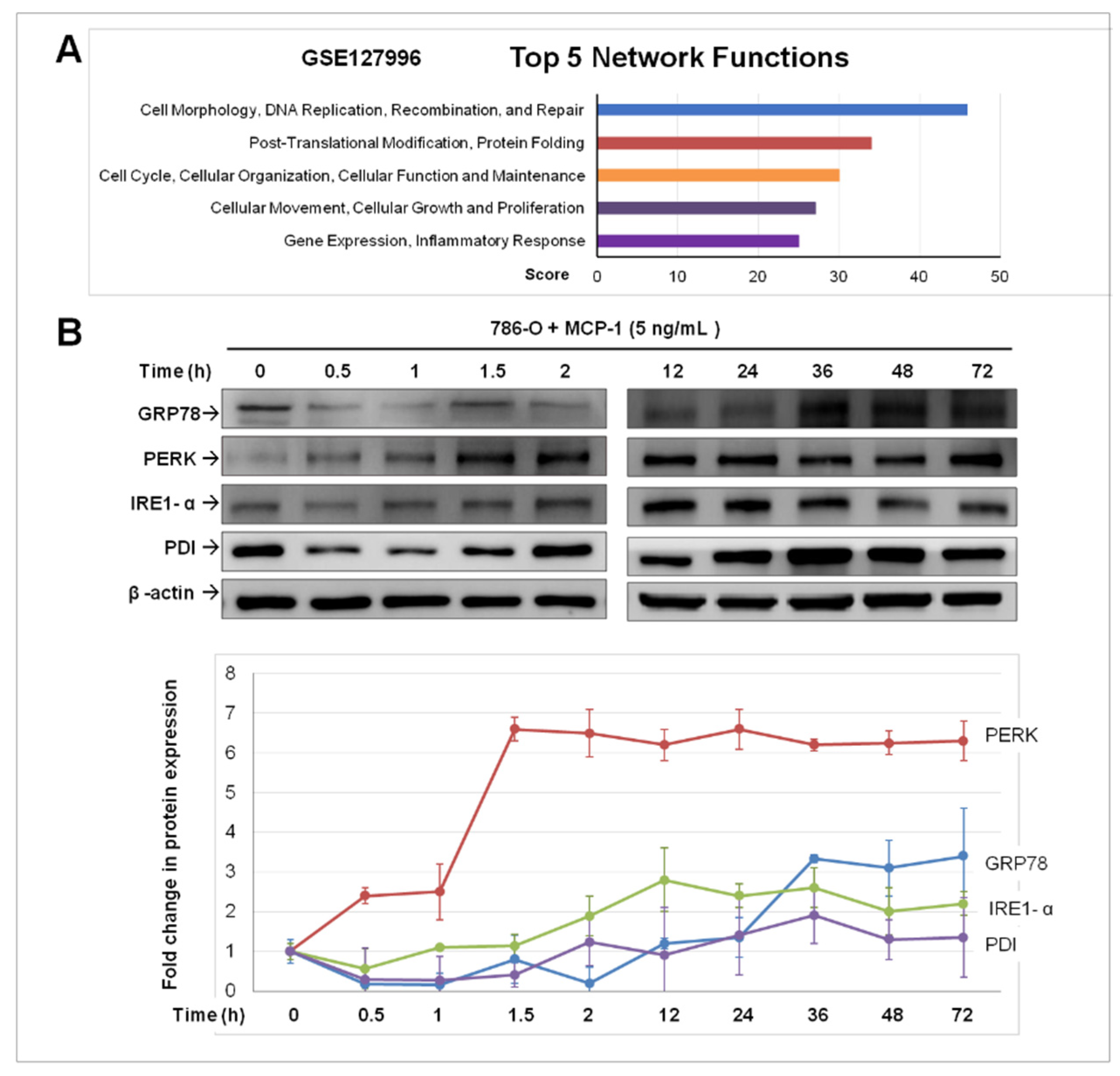
2.3. Treatment of MCP-1 Resulted in Dysregulation of Protein-Folding and Expression of ER Stress Mediators in RCC Cell Line
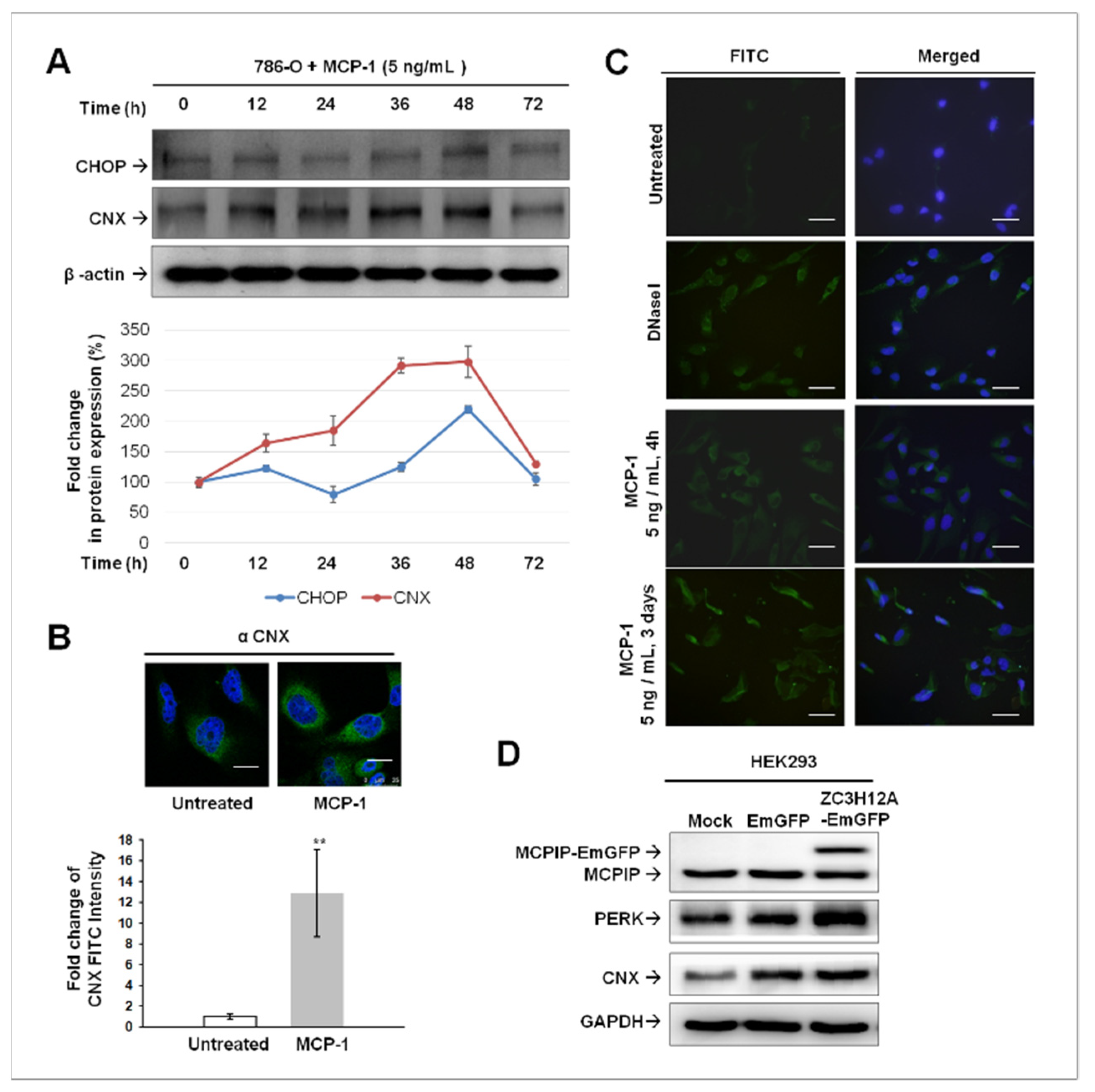
2.4. ER Stress-Mediated Apoptosis Occurred in MCP-1-Treated Cultured 786-O Cells
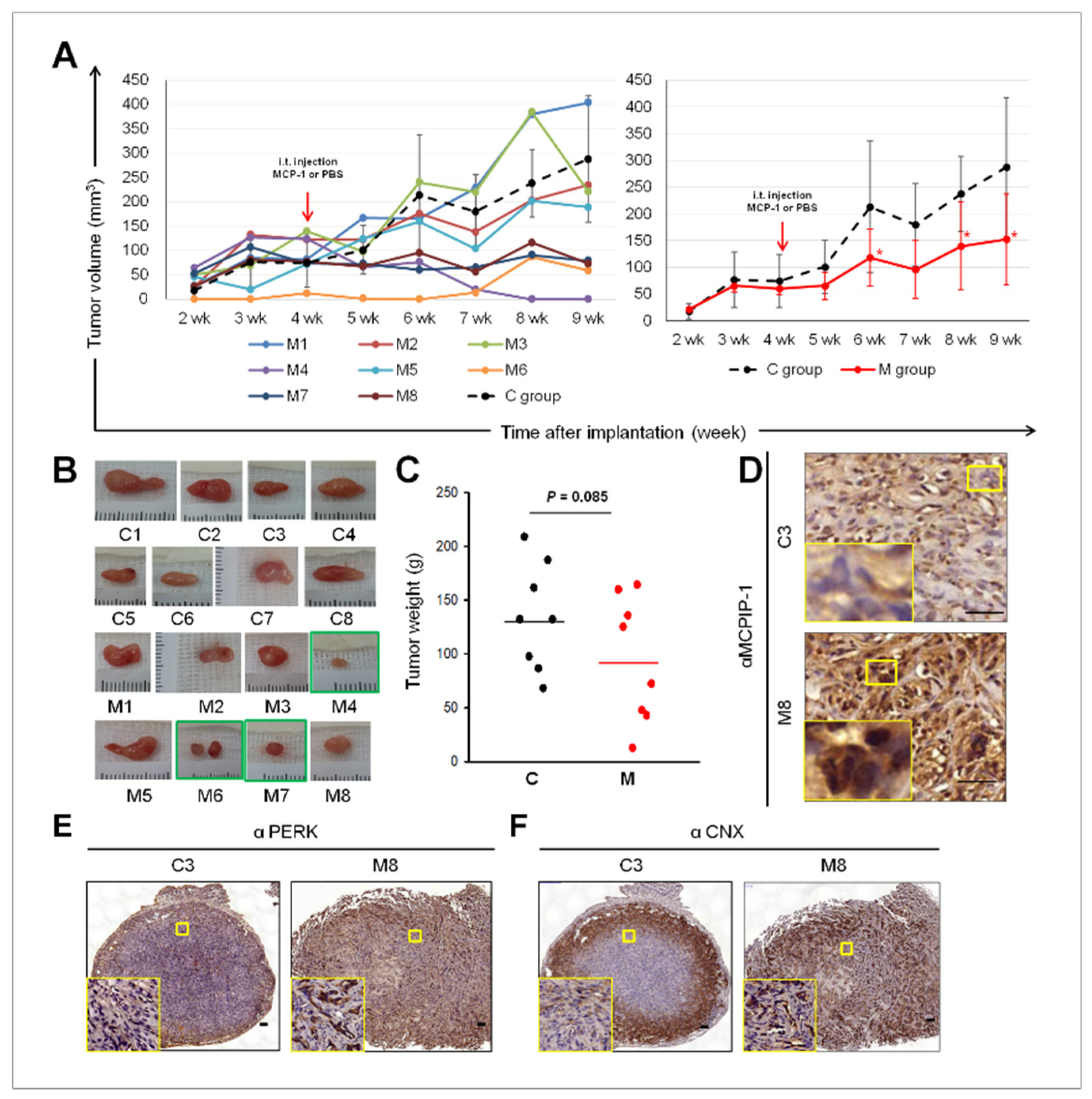
2.5. Intra-Tumoral Injection of MCP-1 Led to Expression of MCPIP-1, PERK, and CNX, as well as Reduced Tumor Size in a Mouse Xenograft Model of RCC
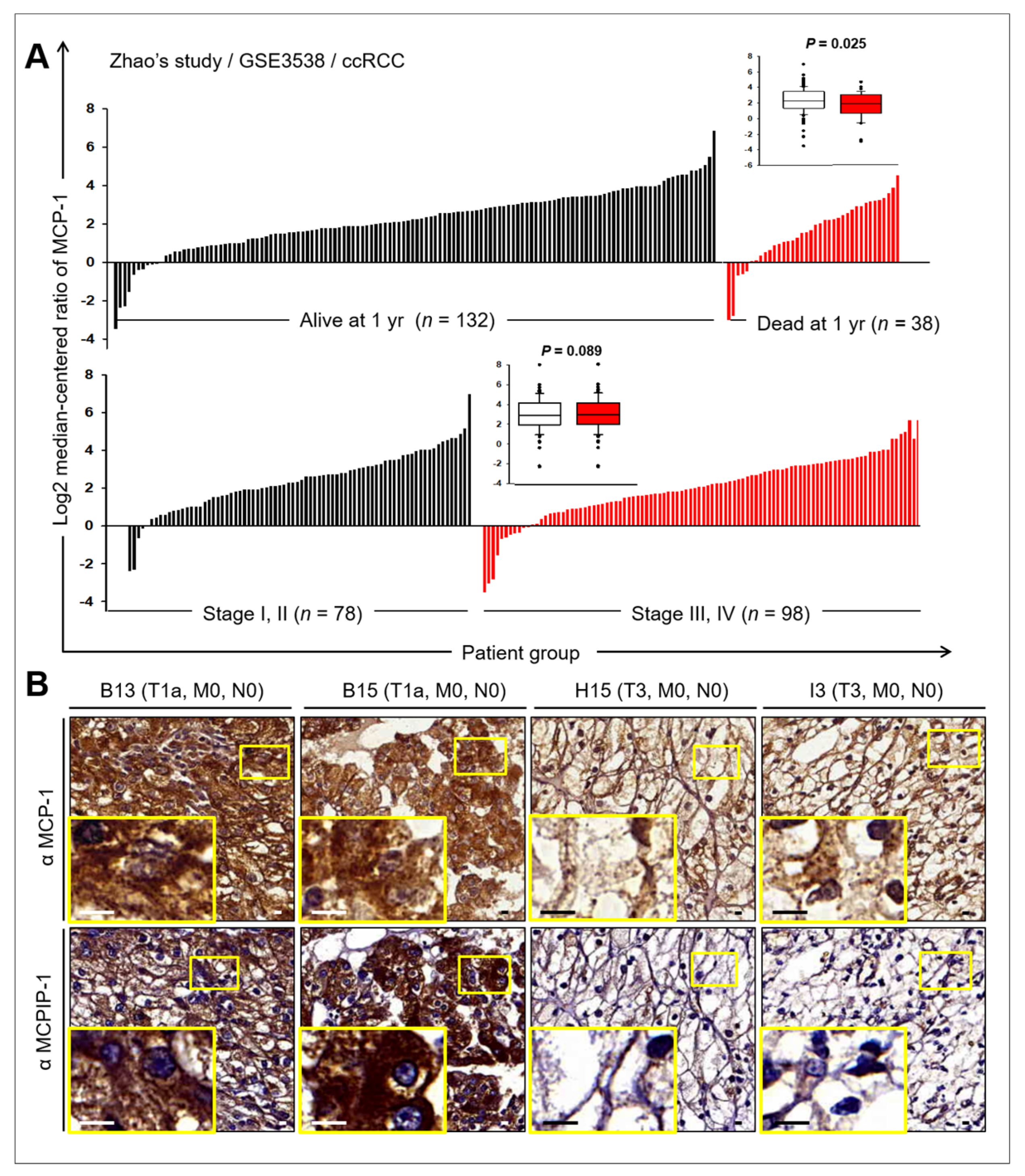
2.6. Prognostic Value of MCP-1 and MCPIP Expression in RCC
3. Discussion
4. Materials and Methods
4.1. Patients and Clinical Samples
4.2. Cell Lines and DNA Transfection
4.3. Chemicals and Reagents
4.4. Western Blot Analysis
4.5. ELISA
4.6. Cytokine Antibody Array Analysis
4.7. Luciferase Reporter Assay
4.8. Quantitative Real-Time qPCR (qPCR)
4.9. Gene Expression Microarray
4.10. Mouse Xenogaft Tumor Model
4.11. Imunohistochemistry (IHC) and Quantitative Staining Measurement of IHC
4.12. TUNEL Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MCP-1 | Monocyte hemoattractant protein-1 |
| MCPIP-1 | MCP-1-induced protein-1 |
| IL-1β | Interleukin-1β |
| RCC | Renal cell carcinoma |
| BiP/GRP | Binding immunoglobulin protein/glucose-regulated protein 78 |
| PERK | RNA-dependent protein kinase-like ER kinase |
| IREα | IRE: inositol requiring element 1α |
| PDI | Protein disulfide isomerase |
| ERO-1α | ER oxidoreductase 1α |
| CHOP | CCAAT enhancer–binding protein homologous protein |
| CNX | calnexin |
| SOCS3 | Suppressor of cytokine signaling-3 |
| NF-κB | Nuclear factor-κB |
| AP-1 | Activator protein 1 |
| ccRCC | Clear cell renal cell carcinoma |
| ER | Endoplasmic reticulum |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay |
| IHC | Immunohistochemistry |
| VHL | von Hippel–Lindau |
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.I.; Lam, J.S.; Figlin, R.A.; Belldegrun, A.S. Surveillance strategies for renal cell carcinoma patients following nephrectomy. Rev. Urol. 2006, 8, 1–7. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, P.; Hudson, M.; Brown, C.; Lord, S.; Gebski, V.; De Souza, P.; Lee, C.K. Markers of systemic inflammation predict survival in patients with advanced renal cell cancer. Br. J. Cancer 2013, 109, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, J.P.; Nayak, B.; Shanmugasundaram, K.; Friedrichs, W.; Sudarshan, S.; Eid, A.A.; DeNapoli, T.; Parekh, D.J.; Gorin, Y.; Block, K. Nox4 mediates renal cell carcinoma cell invasion through hypoxia-induced interleukin 6- and 8- production. PLoS ONE 2012, 7, e30712. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Koguchi, T.; Matsuoka, K.; Onagi, A.; Tanji, R.; Takinami-Honda, R.; Hoshi, S.; Onoda, M.; Kurimura, Y.; Hata, J.; et al. Interleukin-6 induces drug resistance in renal cell carcinoma. Fukushima J. Med. Sci. 2018, 64, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Lu, F.; Liu, C.; Xu, K.; Huang, J.; Yu, D.; Bi, L. IL-8 induces the epithelial-mesenchymal transition of renal cell carcinoma cells through the activation of AKT signaling. Oncol. Lett. 2016, 12, 1915–1920. [Google Scholar] [CrossRef]
- Petrella, B.L.; Vincenti, M.P. Interleukin-1beta mediates metalloproteinase-dependent renal cell carcinoma tumor cell invasion through the activation of CCAAT enhancer binding protein beta. Cancer Med. 2012, 1, 17–27. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, H.; Zhou, L.; Liu, Z.; Fu, H.; Zhu, Y.; Xu, L.; Xu, J. CCL2/CCR2 axis is associated with postoperative survival and recurrence of patients with non-metastatic clear-cell renal cell carcinoma. Oncotarget 2016, 7, 51525–51534. [Google Scholar] [CrossRef] [Green Version]
- Arakaki, R.; Yamasaki, T.; Kanno, T.; Shibasaki, N.; Sakamoto, H.; Utsunomiya, N.; Sumiyoshi, T.; Shibuya, S.; Tsuruyama, T.; Nakamura, E.; et al. CCL2 as a potential therapeutic target for clear cell renal cell carcinoma. Cancer Med. 2016, 5, 2920–2933. [Google Scholar] [CrossRef]
- Brenner, D.R.; Scherer, D.; Muir, K.; Schildkraut, J.; Boffetta, P.; Spitz, M.R.; Le Marchand, L.; Chan, A.T.; Goode, E.L.; Ulrich, C.M.; et al. A review of the application of inflammatory biomarkers in epidemiologic cancer research. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1729–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Voronov, E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol. Rev. 2008, 222, 222–241. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Syu, S.H.; Liu, K.J.; Chu, P.Y.; Yang, W.C.; Lin, P.; Shieh, W.Y. Interleukin-1 beta transactivates epidermal growth factor receptor via the CXCL1-CXCR2 axis in oral cancer. Oncotarget 2015, 6, 38866–38880. [Google Scholar] [CrossRef] [Green Version]
- Younce, C.W.; Kolattukudy, P.E. MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein, MCPIP. Biochem. J. 2010, 426, 43–53. [Google Scholar] [CrossRef]
- Zhou, L.; Azfer, A.; Niu, J.; Graham, S.; Choudhury, M.; Adamski, F.M.; Younce, C.; Binkley, P.F.; Kolattukudy, P.E. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 2006, 98, 1177–1185. [Google Scholar] [CrossRef]
- Küper, C.; Beck, F.X.; Neuhofer, W. Autocrine MCP-1/CCR2 signaling stimulates proliferation and migration of renal carcinoma cells. Oncol. Lett. 2016, 12, 2201–2209. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Delom, F.; Emadali, A.; Cocolakis, E.; Lebrun, J.J.; Nantel, A.; Chevet, E. Calnexin-dependent regulation of tunicamycin-induced apoptosis in breast carcinoma MCF-7 cells. Cell Death Differ. 2007, 14, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Guerin, R.; Arseneault, G.; Dumont, S.; Rokeach, L.A. Calnexin is involved in apoptosis induced by endoplasmic reticulum stress in the fission yeast. Mol. Biol. Cell 2008, 19, 4404–4420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuppini, A.; Groenendyk, J.; Cormack, L.A.; Shore, G.; Opas, M.; Bleackley, R.C.; Michalak, M. Calnexin deficiency and endoplasmic reticulum stress-induced apoptosis. Biochemistry 2002, 41, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ljungberg, B.; Grankvist, K.; Rasmuson, T.; Tibshirani, R.; Brooks, J.D. Gene expression profiling predicts survival in conventional renal cell carcinoma. PLoS Med. 2006, 3, e13. [Google Scholar] [CrossRef]
- Mordente, A.; Meucci, E.; Martorana, G.E.; Silvestrini, A. Cancer Biomarkers Discovery and Validation: State of the Art, Problems and Future Perspectives. Adv. Exp. Med. Biol 2015, 867, 9–26. [Google Scholar]
- Monti, P.; Leone, B.E.; Marchesi, F.; Balzano, G.; Zerbi, A.; Scaltrini, F.; Pasquali, C.; Calori, G.; Pessi, F.; Sperti, C.; et al. The CC chemokine MCP-1/CCL2 in pancreatic cancer progression: Regulation of expression and potential mechanisms of antimalignant activity. Cancer Res. 2003, 63, 7451–7461. [Google Scholar]
- Ozbek, E.; Otunctemur, A.; Sahin, S.; Dursun, M.; Besiroglu, H.; Koklu, I.; Polat, E.C.; Erkoc, M.; Danis, E.; Bozkurt, M. Renal cell carcinoma is more aggressive in Turkish patients with the metabolic syndrome. Asian Pac. J. Cancer Prev. 2013, 14, 7351–7354. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.M.; Zhu, Y.; Ye, D.W. Metabolic syndrome and renal cell carcinoma. World J. Surg. Oncol. 2014, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Ghemrawi, R.; Battaglia-Hsu, S.F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Graff, R.E.; Sanchez, A.; Tobias, D.K.; Rodriguez, D.; Barrisford, G.W.; Blute, M.L.; Li, Y.; Sun, Q.; Preston, M.A.; Wilson, K.M.; et al. Type 2 Diabetes in Relation to the Risk of Renal Cell Carcinoma Among Men and Women in Two Large Prospective Cohort Studies. Diabetes Care 2018, 41, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Van Hemelrijck, M.; Garmo, H.; Hammar, N.; Jungner, I.; Walldius, G.; Lambe, M.; Holmberg, L. The interplay between lipid profiles, glucose, BMI and risk of kidney cancer in the Swedish AMORIS study. Int. J. Cancer 2012, 130, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Wong, T.S.; Chan, J.Y.; Lu, S.C.; Lin, P.; Cheng, A.J.; Chen, Y.J.; Chang, J.S.; Hsiao, S.H.; Leu, Y.W.; et al. Epigenetic regulation of the X-linked tumour suppressors BEX1 and LDOC1 in oral squamous cell carcinoma. J. Pathol. 2013, 230, 298–309. [Google Scholar] [CrossRef]
- Lee, C.H.; Yang, J.R.; Chen, C.Y.; Tsai, M.H.; Hung, P.F.; Chen, S.J.; Chiang, S.L.; Chang, H.; Lin, P. Novel STAT3 Inhibitor LDOC1 Targets Phospho-JAK2 for Degradation by Interacting with LNX1 and Regulates the Aggressiveness of Lung Cancer. Cancers 2019, 11, 63. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Expression | No. of Cases | Percentage |
|---|---|---|
| MCP-1 in tumor tissue | ||
| Negative | 0 | 0 |
| Positive | 75 | 100% |
| Weak | 30 | 40% |
| Moderate | 29 | 38.7% |
| Strong | 16 | 21.3% |
| MCP-1 in adjacent non-tumor tissue | ||
| Negative | 0 | 0 |
| Positive | 75 | 100% |
| Weak | 7 | 9.3% |
| Moderate | 33 | 44% |
| Strong | 35 | 46.7% |
| MCPIP in tumor tissue | ||
| Negative | 17 | 22.7% |
| Positive | 58 | 77.3% |
| Weak | 21 | 28% |
| Moderate | 32 | 42.7% |
| Strong | 5 | 6.7% |
| MCPIP in adjacent non-tumor tissue | ||
| Negative | 10 | 13.3% |
| Positive | 65 | 86.7% |
| Weak | 40 | 53.3% |
| Moderate | 25 | 33.3% |
| Strong | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-H.; Hung, P.-F.; Lu, S.-C.; Chung, H.-L.; Chiang, S.-L.; Wu, C.-T.; Chou, W.-C.; Sun, C.-Y. MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1β in Renal Cell Carcinoma through ER Stress-Mediated Apoptosis. Int. J. Mol. Sci. 2019, 20, 6101. https://doi.org/10.3390/ijms20236101
Lee C-H, Hung P-F, Lu S-C, Chung H-L, Chiang S-L, Wu C-T, Chou W-C, Sun C-Y. MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1β in Renal Cell Carcinoma through ER Stress-Mediated Apoptosis. International Journal of Molecular Sciences. 2019; 20(23):6101. https://doi.org/10.3390/ijms20236101
Chicago/Turabian StyleLee, Chia-Huei, Pin-Feng Hung, Shang-Chieh Lu, Hsuan-Lien Chung, Shang-Lun Chiang, Chun-Te Wu, Wei-Chun Chou, and Chiao-Yin Sun. 2019. "MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1β in Renal Cell Carcinoma through ER Stress-Mediated Apoptosis" International Journal of Molecular Sciences 20, no. 23: 6101. https://doi.org/10.3390/ijms20236101
APA StyleLee, C. -H., Hung, P. -F., Lu, S. -C., Chung, H. -L., Chiang, S. -L., Wu, C. -T., Chou, W. -C., & Sun, C. -Y. (2019). MCP-1/MCPIP-1 Signaling Modulates the Effects of IL-1β in Renal Cell Carcinoma through ER Stress-Mediated Apoptosis. International Journal of Molecular Sciences, 20(23), 6101. https://doi.org/10.3390/ijms20236101