Alternative Strategies to Inhibit Tumor Vascularization


 ,
,  and
and 
Abstract
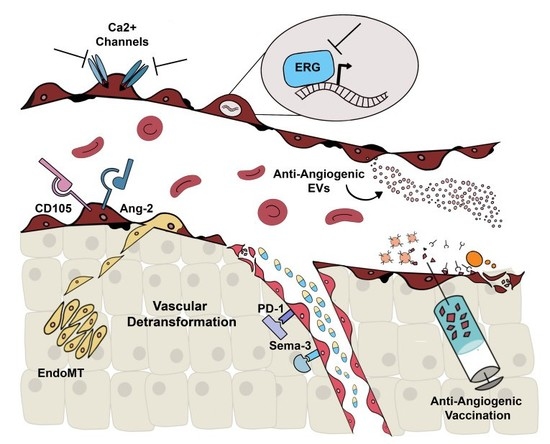
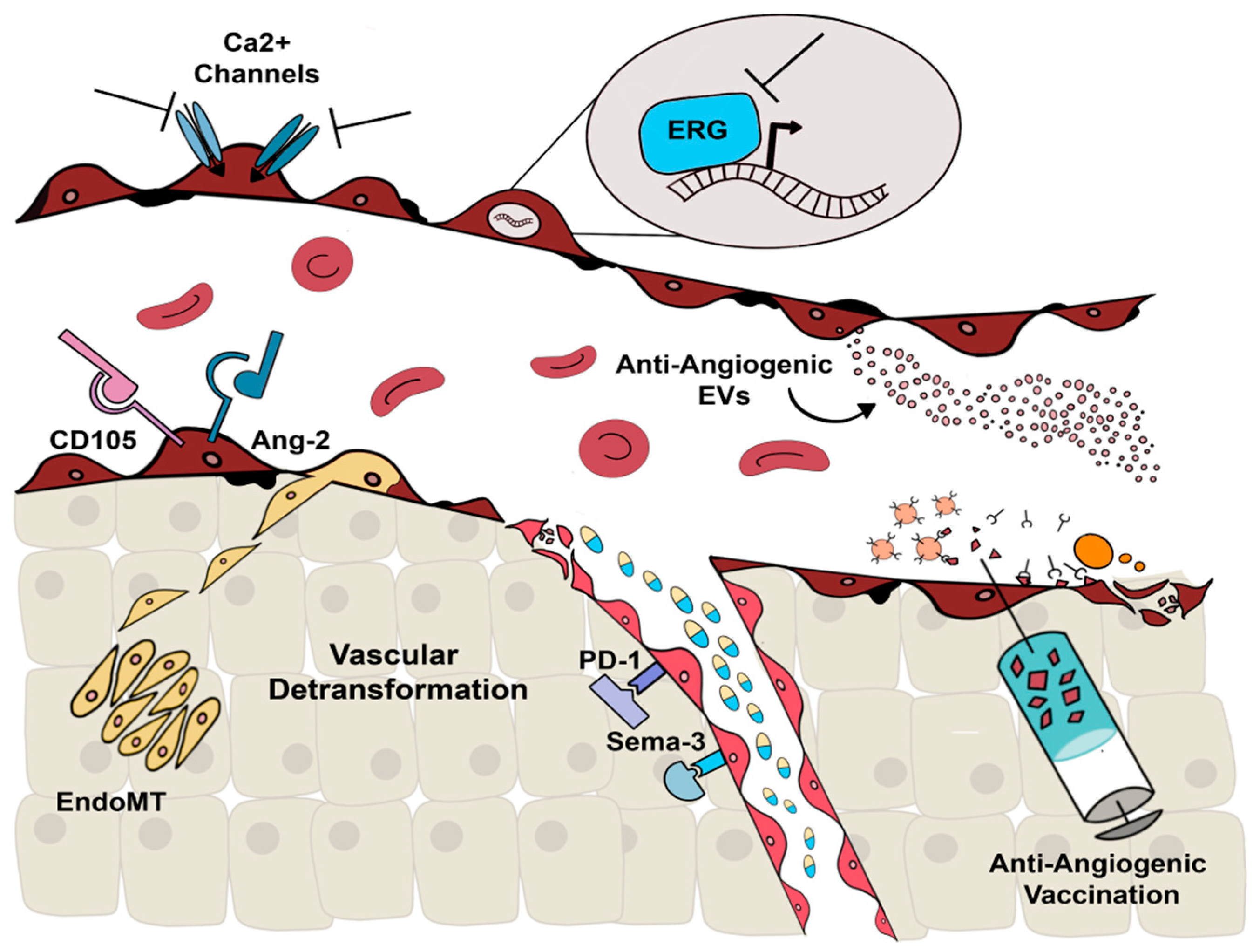
:
1. Introduction
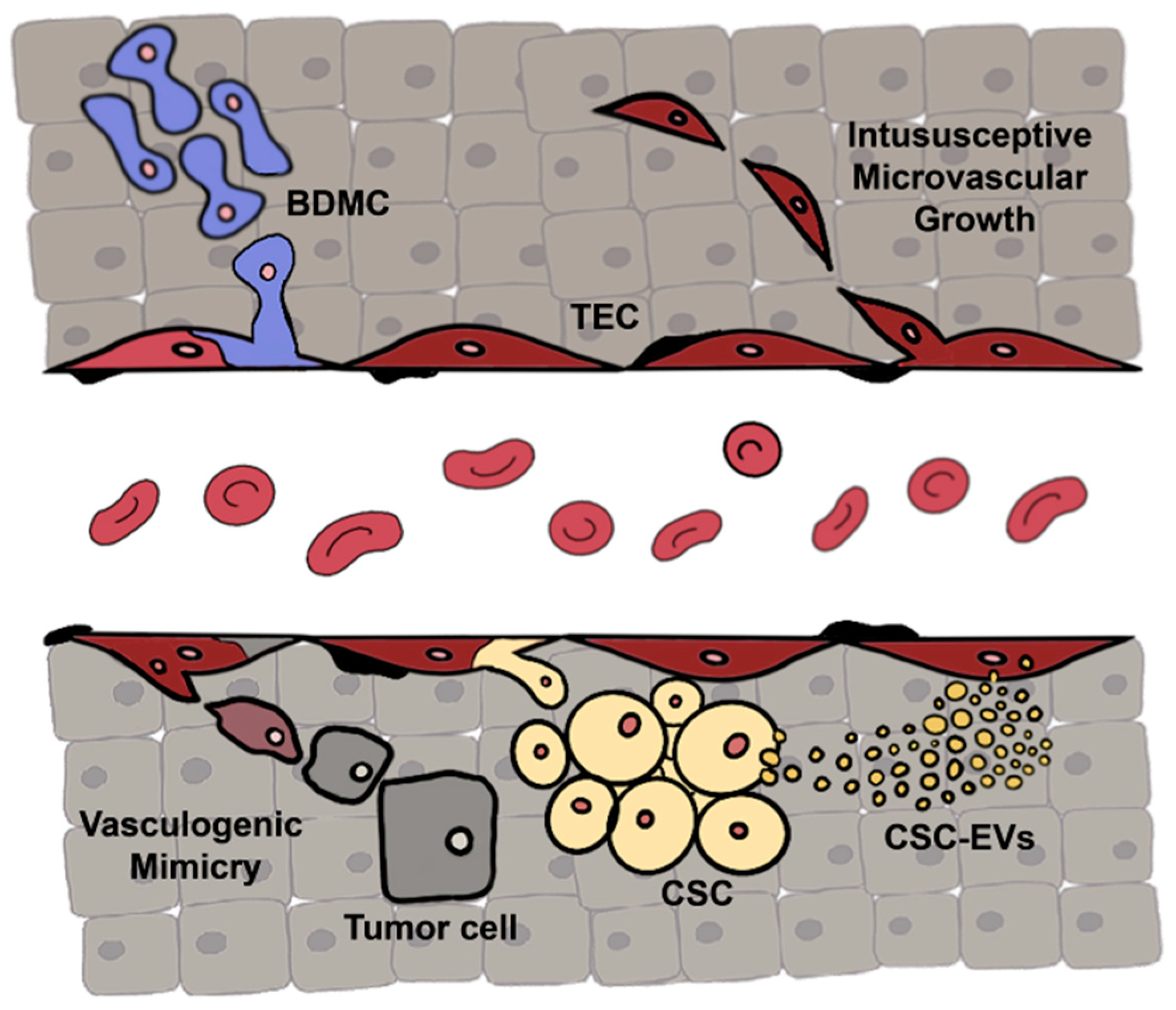
1.1. Tumor Endothelial Cell Characterization
1.2. Tumor Endothelial Cell Origin
1.3. Classic Anti-Angiogenic Therapies
2. Alternative Molecular Targets
2.1. Alternative Anti-Angiogenic Antibodies
2.2. Ca2+-Permeable Channels
2.3. ERG
3. Extracellular Vesicles
4. Anti-Angiogenic Vaccination
5. Vascular Normalization and Detransformation
Funding
Acknowledgments
Conflicts of Interest
References
- Judah Folkman Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 262, 1182–1186.
- De Bock, K.; Cauwenberghs, S.; Carmeliet, P. Vessel abnormalization: Another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr. Opin. Genet. Dev. 2011, 21, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Ugel, S.; Facciponte, J.; Facciabene, A. The dark side of tumor-associated endothelial cells. Semin. Immunol. 2018, 35, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Moon, R.T.; Dev, G.; Cadigan, K.M.; Nusse, R.; Cells, G.; Hart, M.J.; Santos, R.D.L.; Albert, I.N.; Rubinfeld, B.; et al. Genes Expressed in Human Tumor Endothelium. Science 2000, 289, 1197–1202. [Google Scholar]
- Hida, K.; Klagsbrun, M. A new perspective on tumor endothelial cells: Unexpected chromosome and centrosome abnormalities. Cancer Res. 2005, 65, 2507–2510. [Google Scholar] [CrossRef] [Green Version]
- Bussolati, B.; Deambrosis, I.; Russo, S.; Deregibus, M.C.; Camussi, G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003, 17, 1159–1161. [Google Scholar] [CrossRef]
- Pla, A.F.; Brossa, A.; Bernardini, M.; Genova, T.; Grolez, G.; Villers, A.; Leroy, X.; Prevarskaya, N.; Gkika, D.; Bussolati, B. Differential sensitivity of prostate tumor derived endothelial cells to sorafenib and sunitinib. BMC Cancer 2014, 14, 1–13. [Google Scholar]
- Xiong, Y.Q.; Sun, H.C.; Zhang, W.; Zhu, X.D.; Zhuang, P.Y.; Zhang, J.B.; Wang, L.; Wu, W.Z.; Qin, L.X.; Tang, Z.Y. Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef] [Green Version]
- Bussolati, B.; Grange, C.; Camussi, G. Tumor exploits alternative strategies to achieve vascularization. FASEB J. 2011, 25, 2874–2882. [Google Scholar] [CrossRef]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Song, N.; Luo, Y. Role of bone marrow-derived cells in angiogenesis: Focus on macrophages and pericytes. Cancer Microenviron. 2012, 5, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Bussolati, B.; Grange, C.; Collino, F.; Graziano, M.E.; Ferrando, U.; Camussi, G. CD133+ renal progenitor cells contribute to tumor angiogenesis. Am. J. Pathol. 2006, 169, 2223–2235. [Google Scholar] [CrossRef] [Green Version]
- Bussolati, B.; Bruno, S.; Grange, C.; Ferrando, U.; Camussi, G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008, 22, 3696–3705. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–830. [Google Scholar] [CrossRef]
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Luo, H. Overview of advances in vasculogenic mimicry — A potential target for tumor therapy. Cancer Manag. Res. 2018, 10, 2429–2437. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Djonov, V. Intussusceptive microvascular growth in tumors. Cancer Lett. 2012, 316, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, G.; Chirivi, R.G.S.; Fiorentini, S.; Dossi, R.; Bonardelli, S.; Giulini, S.M.; Zanetta, G.; Landoni, F.; Graziotti, P.P.; Turano, A.; et al. Phenotypic and functional characteristics of tumour-derived microvascular endothelial cells. Clin. Exp. Metastasis 1999, 17, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.E.; Oltrogge, J.B.; Von Briesen, H.; Engelhardt, B.; Woelki, U.; Schlote, W.; Lorenz, R.; Bratzke, H.; Kirkpatrick, C.J. Isolation and molecular characterization of brain microvascular endothelial cells from human brain tumors. Vitr. Cell. Dev. Biol. Anim. 2002, 38, 273–281. [Google Scholar] [CrossRef]
- Allport, J.R.; Weissleder, R. Murine Lewis lung carcinoma-derived endothelium expresses markers of endothelial activation and requires tumor-specific extracellular matrix in vitro. Neoplasia 2003, 5, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Streubel, B.; Chott, A.; Huber, D.; Exner, M.; Jäger, U.; Wagner, O.; Schwarzinger, I. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N. Engl. J. Med. 2004, 351, 250–259. [Google Scholar] [CrossRef]
- Hida, K.; Hida, Y.; Amin, D.N.; Flint, A.F.; Panigrahy, D.; Morton, C.C.; Klagsbrun, M. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004, 64, 8249–8255. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Bussolati, B.; Bruno, S.; Fonsato, V.; Sapino, A.; Camussi, G. Isolation and characterization of human breast tumor-derived endothelial cells. Oncol. Rep. 2006, 15, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Amin, D.N.; Hida, K.; Bielenberg, D.R.; Klagsbrun, M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006, 66, 2173–2180. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.Q.; Zhang, W.J.; Niu, J.X.; Ye, L.Y.; Yang, Z.H.; Grau, G.E.; Lou, J.N. Phenotypic and functional differences between human liver cancer endothelial cells and liver sinusoidal endothelial cells. J. Vasc. Res. 2007, 45, 78–86. [Google Scholar] [CrossRef]
- Buckanovich, R.J.; Sasaroli, D.; O’Brien-Jenkins, A.; Botbyl, J.; Hammond, R.; Katsaros, D.; Sandaltzopoulos, R.; Liotta, L.A.; Gimotty, P.A.; Coukos, G. Tumor vascular proteins as biomarkers in ovarian cancer. J. Clin. Oncol. 2007, 25, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Bonome, T.; Li, Y.; Kamat, A.A.; Han, L.Y.; Schmandt, R.; Coleman, R.L.; Gershenson, D.M.; Jaffe, R.B.; Birrer, M.J.; et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007, 67, 1757–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Wu, M.; Chen, Y.; Xu, G.; Wei, J.; Li, Q.; Song, A.; Zhao, L.; Li, S.; Han, Z.; et al. Isolation and characterization of lymphatic endothelial cells from human glossal lymphangioma. Oncol. Rep. 2010, 25, 223–230. [Google Scholar]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jayson, G.C.; Hicklin, D.J.; Ellis, L.M. Antiangiogenic therapy-evolving view based on clinical trial results. Nat. Rev. Clin. Oncol. 2012, 9, 297–303. [Google Scholar] [CrossRef]
- McIntyre, A.; Harris, A.L. Metabolic and hypoxic adaptation to anti—angiogenic therapy: A target for induced essentiality. EMBO Mol. Med. 2015, 7, 368–379. [Google Scholar] [CrossRef]
- Mancuso, M.R.; Davis, R.; Norberg, S.M.; Brien, S.O.; Sennino, B.; Nakahara, T.; Yao, V.J.; Inai, T.; Brooks, P.; Freimark, B.; et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Investig. 2006, 116, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Hainsworth, J.D.; Spigel, D.R.; Sosman, J.A.; Burris, H.A.; Farley, C.; Cucullu, H.; Yost, K.; Hart, L.L.; Sylvester, L.; Waterhouse, D.M.; et al. Treatment of advanced renal cell carcinoma with the combination bevacizumab/erlotinib/imatinib: A phase I/II trial. Clin. Genitourin. Cancer 2007, 5, 427–432. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.H.; Kim, M.R.; Jang, J.H.; Na, H.J.; Lee, S. A review of anti-angiogenic targets for monoclonal antibody cancer therapy. Int. J. Mol. Sci. 2017, 18, 1786. [Google Scholar] [CrossRef] [Green Version]
- McNeel, D.G.; Eickhoff, J.; Lee, F.T.; King, D.M.; Alberti, D.; Thomas, J.P.; Friedl, A.; Kolesar, J.; Marnocha, R.; Volkman, J.; et al. Phase I trial of a monoclonal antibody specific for α v β 3 integrin (MEDI-522) in patients with advanced malignancies, including an assessment of effect on tumor perfusion. Clin. Cancer Res. 2005, 11, 7851–7860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binz, H.K.; Bakker, T.R.; Phillips, D.J.; Cornelius, A.; Zitt, C.; Göttler, T.; Sigrist, G.; Fiedler, U.; Ekawardhani, S.; Dolado, I.; et al. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin® drug candidate. MAbs 2017, 9, 1262–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, U.; Ekawardhani, S.; Cornelius, A.; Gilboy, P.; Bakker, T.R.; Dolado, I.; Stumpp, M.T.; Dawson, K.M. MP0250, a VEGF and HGF neutralizing DARPin® molecule shows high anti-tumor efficacy in mouse xenograft and patient-derived tumor models. Oncotarget 2017, 8, 98371–98383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, L.; Veirman, K.; de Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Piao, Y.; Jeong, K.J.; Dong, J.; de Groot, J.F. Periostin (POSTN) regulates tumor resistance to antiangiogenic therapy in glioma models. Physiol. Behav. 2016, 176, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Paauwe, M.; Heijkants, R.C.; Oudt, C.H.; Van Pelt, G.W.; Cui, C.; Theuer, C.P.; Hardwick, J.C.H.; Sier, C.F.M.; Hawinkels, L.J.A.C. Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene 2016, 35, 4069–4079. [Google Scholar] [CrossRef]
- Derbyshire, E.J.; Gazdar, A.F.; King, S.W.; Thorpe, P.E.; Derbyshire, E.J.; King, S.W.; Thorpe, P.E.; Gazdar, A.F.; Vitetta, E.S.; Tazzari, P.L.; et al. Up-Regulation of Endoglin on Vascular Endothelial Cells in Human Solid Tumors: Implications for Diagnosis and Therapy. Clin. Cancer Res. 1995, 1, 1623–1634. [Google Scholar]
- Hu, J.; Guan, W.; Liu, P.; Dai, J.; Tang, K.; Xiao, H.; Qian, Y.; Sharrow, A.C.; Ye, Z.; Wu, L.; et al. Endoglin Is Essential for the Maintenance of Self-Renewal and Chemoresistance in Renal Cancer Stem Cells. Stem Cell Reports 2017, 9, 464–477. [Google Scholar] [CrossRef] [Green Version]
- Madhav, A.; Andres, A.; Duong, F.; Mishra, R.; Haldar, S.; Liu, Z.; Angara, B.; Gottlieb, R.; Zumsteg, Z.S.; Bhowmick, N.A. Antagonizing CD105 enhances radiation sensitivity in prostate cancer. Oncogene 2018, 37, 4385–4397. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, B.; Zhang, H.; Li, H. Human epithelial ovarian cancer cells expressing cd105, cd44 and cd106 surface markers exhibit increased invasive capacity and drug resistance. Oncol. Lett. 2019, 17, 5351–5360. [Google Scholar] [CrossRef] [Green Version]
- Brossa, A.; Buono, L.; Bussolati, B. Effect of the monoclonal antibody TRC105 in combination with Sunitinib on renal tumor derived endothelial cells. Oncotarget 2018, 9, 22680–22692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, C.R.; Liu, L.; Theuer, C. An adaptive population enrichment phase III trial of TRC105 and pazopanib versus pazopanib alone in patients with advanced angiosarcoma (TAPPAS trial). Ann. Oncol. 2019, 30, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Worley, M.J.; Elias, K.M.; Horowitz, N.S.; Quade, B.J.; Berkowitz, R.S. Durable remission for a woman with refractory choriocarcinoma treated with anti-endoglin monoclonal antibody and bevacizumab: A case from the New England Trophoblastic Disease Center, Brigham and Women’s Hospital and Dana-Farber Cancer Institute. Gynecol. Oncol. 2018, 148, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ma, C.; Ulahannan, S.V.; Rahma, O.E.; Makarova-Rusher, O.; Cao, L.; Yu, Y.; Kleiner, D.E.; Trepel, J.B.; Lee, M.-J.; et al. Phase I and preliminary Phase II study of TRC105 in combination with Sorafenib in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 4633–4641. [Google Scholar] [CrossRef] [Green Version]
- Iamshanova, O.; Fiorio Pla, A.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Munaron, L.; Arcangeli, A. Editorial: Ion fluxes and cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 1–3. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Munaron, L.; Genova, T.; Avanzato, D.; Antoniotti, S.; Pla, A.F. Targeting Calcium Channels to Block Tumor Vascularization. Recent Pat. Anticancer. Drug Discov. 2013, 8, 27–37. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Guerra Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [Green Version]
- Farfariello, V.; Iamshanova, O.; Germain, E.; Fliniaux, I.; Prevarskaya, N. Calcium homeostasis in cancer: A focus on senescence. Biochim. Biophys. Acta 2015, 1853, 1974–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, Y.; Nakajima, H.; Wakayama, Y.; Muto, A.; Kawakami, K.; Fukuhara, S.; Mochizuki, N. Endothelial Ca2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. Elife 2015, 4, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 2016, 9, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, A.M.; Kurusamy, S.; Chen, Y.; Jiang, Z.; Chhabria, K.; MacDonald, R.B.; Kim, H.R.; Wilson, H.L.; van Eeden, F.J.M.; Armesilla, A.L.; et al. Tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar]
- Smani, T.; Gómez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.-M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Pocock, T.M.; Foster, R.R.; Bates, D.O. Evidence of a role for TRPC channels in VEGF-mediated increased vascular permeability in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1015–H1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.-W.; James, A.F.; Foster, R.R.; Hancox, J.C.; Bates, D.O. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1768–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Grange, C.; Antoniotti, S.; Tomatis, C.; Merlino, A.; Bussolati, B.; Munaron, L. Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 2008, 6, 535–545. [Google Scholar] [PubMed] [Green Version]
- Fiorio Pla, A.; Genova, T.; Pupo, E.; Tomatis, C.; Genazzani, A.; Zaninetti, R.; Munaron, L. Multiple roles of protein kinase a in arachidonic acid-mediated Ca2+ entry and tumor-derived human endothelial cell migration. Mol. Cancer Res. 2010, 8, 1466–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, E.; Pla, A.F.; Avanzato, D.; Moccia, F.; Cruz, J.-E.E.A.; Tanzi, F.; Merlino, A.; Mancardi, D.; Munaron, L. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic. Biol. Med. 2011, 51, 1765–1773. [Google Scholar] [CrossRef]
- Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Vieira, J.R.A.; Bussolati, B.; Pla, A.F.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. [Google Scholar] [CrossRef] [Green Version]
- Adapala, R.K.; Thoppil, R.J.; Ghosh, K.; Cappelli, H.C.; Dudley, A.C.; Paruchuri, S.; Keshamouni, V.; Klagsbrun, M.; Meszaros, J.G.; Chilian, W.M.; et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fiorio Pla, A.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a nonchannel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers. 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, E.S.P.; Rao, V.N.; Papas, T.S. The erg gene: A human gene related to the ets oncogene. Proc. Natl. Acad. Sci. USA 1987, 84, 6131–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.V.; Birdsey, G.M.; Randi, A.M. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul. Pharmacol. 2016, 86, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birdsey, G.M.; Dryden, N.H.; Amsellem, V.; Gebhardt, F.; Sahnan, K.; Haskard, D.O.; Dejana, E.; Mason, J.C.; Randi, A.M. Transcription factor erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood 2008, 111, 3498–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birdsey, G.M.; Shah, A.V.; Dufton, N.; Reynolds, L.E.; Almagro, L.O.; Yang, Y.; Aspalter, I.M.; Khan, S.T.; Mason, J.C.; Dejana, E.; et al. The endothelial transcription factor erg promotes vascular stability and growth through Wnt/β-catenin signaling. Dev. Cell 2015, 32, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef]
- Brenner, J.C.; Ateeq, B.; Chinnaiyan, A.M.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic Rationale for Inhibition of Poly ( ADP-Ribose ) Polymerase in ETS Gene Fusion- Positive Prostate Cancer. Cancer Cell. 2011, 23, 664–678. [Google Scholar]
- Björkman, M.; Iljin, K.; Halonen, P.; Sara, H.; Kaivanto, E.; Nees, M.; Kallioniemi, O.P. Defining the molecular action of HDAC inhibitors and synergism with androgen deprivation in ERG-positive prostate cancer. Int. J. Cancer 2008, 123, 2774–2781. [Google Scholar] [CrossRef]
- Nagai, N.; Ohguchi, H.; Nakaki, R.; Matsumura, Y.; Kanki, Y.; Sakai, J.; Aburatani, H.; Minami, T. Downregulation of ERG and FLI1 expression in endothelial cells triggers endothelial-to-mesenchymal transition. PLoS Genet. 2018, 14, 1007826. [Google Scholar] [CrossRef]
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Üren, A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion. PLoS ONE 2011, 6, e19343. [Google Scholar] [CrossRef] [Green Version]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kollipara, R.K.; Srivastava, N.; Li, R.; Ravindranathan, P.; Hernandez, E.; Freeman, E.; Humphries, C.G.; Kapur, P.; Lotan, Y.; et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4251–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Khawar, M.B.; Abbasi, M.H.; Siddique, Z.; Arif, A.; Sheikh, N. An Update on Novel Therapeutic Warfronts of Extracellular Vesicles (EVs) in Cancer Treatment: Where We Are Standing Right Now and Where to Go in the Future. Oxid. Med. Cell. Longev. 2019, 2019, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Dagur, R.S.; Liao, K.; Peeples, E.S.; Hu, G.; Periyasamy, P.; Buch, S. Strategies for the use of Extracellular Vesicles for the Delivery of Therapeutics. J. Neuroimmune Pharmacol. 2019, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Brossa, A.; Fonsato, V.; Bussolati, B. Anti-tumor activity of stem cell-derived extracellular vesicles. Oncotarget 2019, 10, 1872–1873. [Google Scholar] [CrossRef] [PubMed]
- Lopatina, T.; Grange, C.; Fonsato, V.; Tapparo, M.; Brossa, A.; Fallo, S.; Pitino, A.; Herrera-Sanchez, M.B.; Kholia, S.; Camussi, G.; et al. Extracellular vesicles from human liver stem cells inhibit tumor angiogenesis. Int. J. Cancer 2019, 144, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Park, S.R.; Jung, B.K.; Jeon, Y.K.; Lee, Y.S.; Kim, M.K.; Kim, Y.G.; Jang, J.Y.; Kim, C.W. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE 2013, 8, 84256. [Google Scholar] [CrossRef] [Green Version]
- Grigorian-Shamagian, L.; Fereydooni, S.; Liu, W.; Echavez, A.; Marbán, E. Harnessing the heart’s resistance to malignant tumors: Cardiacderived extracellular vesicles decrease fibrosarcoma growth and leukemia-related mortality in rodents. Oncotarget 2017, 8, 99624–99636. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.C.; Ichim, T.E.; Ma, H.; Szymanski, J.; Perez, J.A.; Lopez, J.; Bogin, V.; Patel, A.N.; Marincola, F.M.; Kesari, S. Cancer anti-angiogenesis vaccines: Is the tumor vasculature antigenically unique? J. Transl. Med. 2015, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, M.; Ohsawa, R.; Tsunoda, T.; Hirono, S.; Kawai, M.; Tani, M.; Nakamura, Y.; Yamaue, H. Phase I clinical trial using peptide vaccine for human vascular endothelial growth factor receptor 2 in combination with gemcitabine for patients with advanced pancreatic cancer. Cancer Sci. 2010, 101, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Arao, T.; Miyazaki, M.; Satoh, T.; Okamoto, K.; Tsunoda, T.; Nishio, K.; Nakagawa, K. Clinical phase I study of elpamotide, a peptide vaccine for vascular endothelial growth factor receptor 2, in patients with advanced solid tumors. Cancer Sci. 2012, 103, 2135–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Kurata, T.; Fujisaka, Y.; Kawakami, H.; Tanaka, K.; Okabe, T.; Takeda, M.; Satoh, T.; Yoshida, K.; Tsunoda, T.; et al. Phase I trial of OTS11101, an anti-angiogenic vaccine targeting vascular endothelial growth factor receptor 1 in solid tumor. Cancer Sci. 2013, 104, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Fukuhara, M.; Yamaura, T.; Mutoh, S.; Okabe, N.; Yaginuma, H.; Hasegawa, T.; Yonechi, A.; Osugi, J.; Hoshino, M.; et al. Multiple therapeutic peptide vaccines consisting of combined novel cancer testis antigens and anti-angiogenic peptides for patients with non-small cell lung cancer. J. Transl. Med. 2013, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia Verdecia, B.; Neninger, E.; De La Torre, A.; Leonard, I.; Martínez, R.; Viada, C.; González, G.; Mazorra, Z.; Lage, A.; Crombet, T. Effective inhibition of the epidermal growth factor/epidermal growth factor receptor binding by anti-epidermal growth factor antibodies is related to better survival in advanced non-small-cell lung cancer patients treated with the epidermal growth factor. Clin. Cancer Res. 2008, 14, 840–846. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Tsuno, N.H.; Fujii, T.; Todo, T.; Saito, N.; Takahashi, K. Human umbilical vein endothelial cell vaccine therapy in patients with recurrent glioblastoma. Cancer Sci. 2013, 104, 200–205. [Google Scholar] [CrossRef]
- Ichim, T.E.; Li, S.; Ma, H.; Yurova, Y.V.; Szymanski, J.S.; Patel, A.N.; Kesari, S.; Min, W.P.; Wagner, S.C. Induction of tumor inhibitory anti-angiogenic response through immunization with interferon Gamma primed placental endothelial cells: ValloVaxTM. J. Transl. Med. 2015, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gavilondo, J.V.; Hernández-Bernal, F.; Ayala-Ávila, M.; de la Torre, A.V.; de la Torre, J.; Morera-Díaz, Y.; Bequet-Romero, M.; Sánchez, J.; Valenzuela, C.M.; Martín, Y.; et al. Specific active immunotherapy with a VEGF vaccine in patients with advanced solid tumors. Results of the CENTAURO antigen dose escalation phase I clinical trial. Vaccine 2014, 32, 2241–2250. [Google Scholar] [CrossRef]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef]
- Yuan, F.; Chen, Y.; Dellian, M.; Safabakhsh, N.; Ferrara, N.; Jain, R.K. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc. Natl. Acad. Sci. USA 1996, 93, 14765–14770. [Google Scholar] [CrossRef] [Green Version]
- Arjaans, M.; Schröder, C.P.; Oosting, S.F.; Dafni, U.; Kleibeuker, J.E.; De Vries, E.G.E. VEGF pathway targeting agents, vessel normalization and tumor drug uptake: From bench to bedside. Oncotarget 2016, 7, 21247–21258. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Serini, G.; Bussolino, F.; Maione, F.; Giraudo, E. Class 3 semaphorins: Physiological vascular normalizing agents for anti-cancer therapy. J. Intern. Med. 2013, 273, 138–155. [Google Scholar] [CrossRef] [Green Version]
- Maione, F.; Molla, F.; Meda, C.; Latini, R.; Zentilin, L.; Giacca, M.; Seano, G.; Serini, G.; Bussolino, F.; Giraudo, E. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J. Clin Invest. 2009, 119, 3356–3372. [Google Scholar] [CrossRef]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual PD --1 and VEGFR —2 blockade promotes vascular normalization and enhances anti--tumor immune responses in HCC. Hepatology 2019, 0–2. [Google Scholar] [CrossRef]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [Green Version]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Rmili, C.W.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, 9670. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 12, 385–413. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y. Vascular Detransformation for Cancer Therapy. Trends in Cancer 2019, 5, 1–4. [Google Scholar] [CrossRef]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 1–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Species | Year | References |
|---|---|---|---|
| Glioblastoma | Human | 1999 | Alessandri et al. [23] |
| Colon | Human | 2000 | St. Croix et al. [5] |
| Brain tumors | Human | 2002 | Unger et al. [24] |
| Renal | Human | 2003 | Bussolati et al. [7] |
| Lung | Mouse | 2003 | Allport et al. [25] |
| B-Cell lymphoma | Human | 2004 | Streubel et al. [26] |
| Liposarcoma and melanoma | Mouse | 2004 | Hida et al. [27] |
| Breast | Human | 2006 | Grange et al. [28] |
| Breast | Mouse | 2006 | Amin et al. [29] |
| Liver | Human | 2007 | Wu et al. [30] |
| Ovary | Human | 2007 | Buckanovitch et al. [31] Lu et al. [32] |
| Glossal lymphangioma | Human | 2010 | You et al. [33] |
| Prostate | Human | 2014 | Fiorio et al. [8] |
| Drug Name | Type | Targets | Tumor Type | Combined Therapy |
|---|---|---|---|---|
| Bevacizumab | mAb | VEGF-A | Colorectal, lung, glioblastoma, renal cell carcinoma, breast, brain, ovarian, cervical, fallopian tube, and peritoneal cancer | Fluoropirimidine, Cisplatinum, Paclitaxel, Interferon a-2a |
| Sorafenib | TKI | VEGFR1/2/3, PDGFR, c-kit | Renal cell carcinoma, liver, thyroid, desmoid tumors | |
| Sunitinib | TKI | VEGFR1/2/3, PDGFR, c-kit, FLT-3, Ret | Renal cell carcinoma, gastrointestinal stromal, pancreatic neuroendocrine cancer, and leukemia | |
| Pazopanib | TKI | VEGFR1/2/3, PDGFR, c-kit, FGFR | Renal cell carcinoma and soft tissue sarcoma | |
| Axitinib | TKI | VEGFR1/2/3, c-kit, PDGFR | Renal cell carcinoma | |
| Regorafenib | TKI | VEGFR1/2/3, PDGFRα/β, FGFR1/2, Tie2, c-Kit | Metastatic colorectal cancer, advanced gastrointestinal stromal cancer and advanced hepatocellular carcinoma | |
| Cabozantinib | TKI | c-MET, VEGFR2, AXL, Ret | Medullary thyroid cancer and renal cell carcinoma | |
| Nintedanib | TKI | VEGFR1/2/3, PDGFR, FLT-3 | Idiopatic pulmonary fibrosis, lung cancer | Docetaxel |
| Levantinib | TKI | VEGFR1/2/3, PDGFR, FGFR, Ret, c-Kit | Thyroid cancer and renal cell carcinoma | Everolimus |
| Vandetanib | TKI | VEGFR1/2/3, EGFR, and Ret | Medullary thyroid cancer |
| Antigens | Vaccine Type | Tumor Type | Phase | REF/NIH N. |
|---|---|---|---|---|
| VEGF-A | Recombinant human VEGF-A-121 isoform | Advanced solid tumors | I | Gavilondo 2014 [108] |
| VEGFRs | VEGFR2-169 peptide | Pancreatic cancer | I | Miyazawa 2010 [101] |
| VEGFR1-1084 and VEGFR2-169 peptides | I/II | NCT00655785 | ||
| VEGFR1-A2-770 peptide | I/II | NCT00683085 | ||
| VEGFR2-169 peptide | Advanced solid tumors | I | Okamoto 2012 [102] | |
| VEGFR1-1084 peptide | I | Hayashi 2013 [103] | ||
| VEGFR2, VEGFR1, URLC10, TTK, CDCA1 multipeptide | Non small cell lung cancer | I | Suzuki 2013 [104] | |
| Survivin | hTERT/survivin/CMV multipeptide | Breast cancer | I | NCT01660529 |
| Survivin long peptide | Neuroendocrine tumors | I | NCT03879694 | |
| Salmonella-based Survivin peptide | Multiple myeloma | I/II | NCT03762291 | |
| EGF | Recombinant Human EGF-rP64K/Montanide ISA 51 peptide | Non-small cell lung cancer | II | Garcia 2008 [105] |
| II/III | NCT00516685 | |||
| III | NCT02187367 | |||
| III | NCT01444118 | |||
| Non-small cell lung cancer, squamous head and neck cancer | I/II | NCT02955290 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brossa, A.; Buono, L.; Fallo, S.; Fiorio Pla, A.; Munaron, L.; Bussolati, B. Alternative Strategies to Inhibit Tumor Vascularization. Int. J. Mol. Sci. 2019, 20, 6180. https://doi.org/10.3390/ijms20246180
Brossa A, Buono L, Fallo S, Fiorio Pla A, Munaron L, Bussolati B. Alternative Strategies to Inhibit Tumor Vascularization. International Journal of Molecular Sciences. 2019; 20(24):6180. https://doi.org/10.3390/ijms20246180
Chicago/Turabian StyleBrossa, Alessia, Lola Buono, Sofia Fallo, Alessandra Fiorio Pla, Luca Munaron, and Benedetta Bussolati. 2019. "Alternative Strategies to Inhibit Tumor Vascularization" International Journal of Molecular Sciences 20, no. 24: 6180. https://doi.org/10.3390/ijms20246180
APA StyleBrossa, A., Buono, L., Fallo, S., Fiorio Pla, A., Munaron, L., & Bussolati, B. (2019). Alternative Strategies to Inhibit Tumor Vascularization. International Journal of Molecular Sciences, 20(24), 6180. https://doi.org/10.3390/ijms20246180