Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics

 ,
,  ,
,  ,
, 
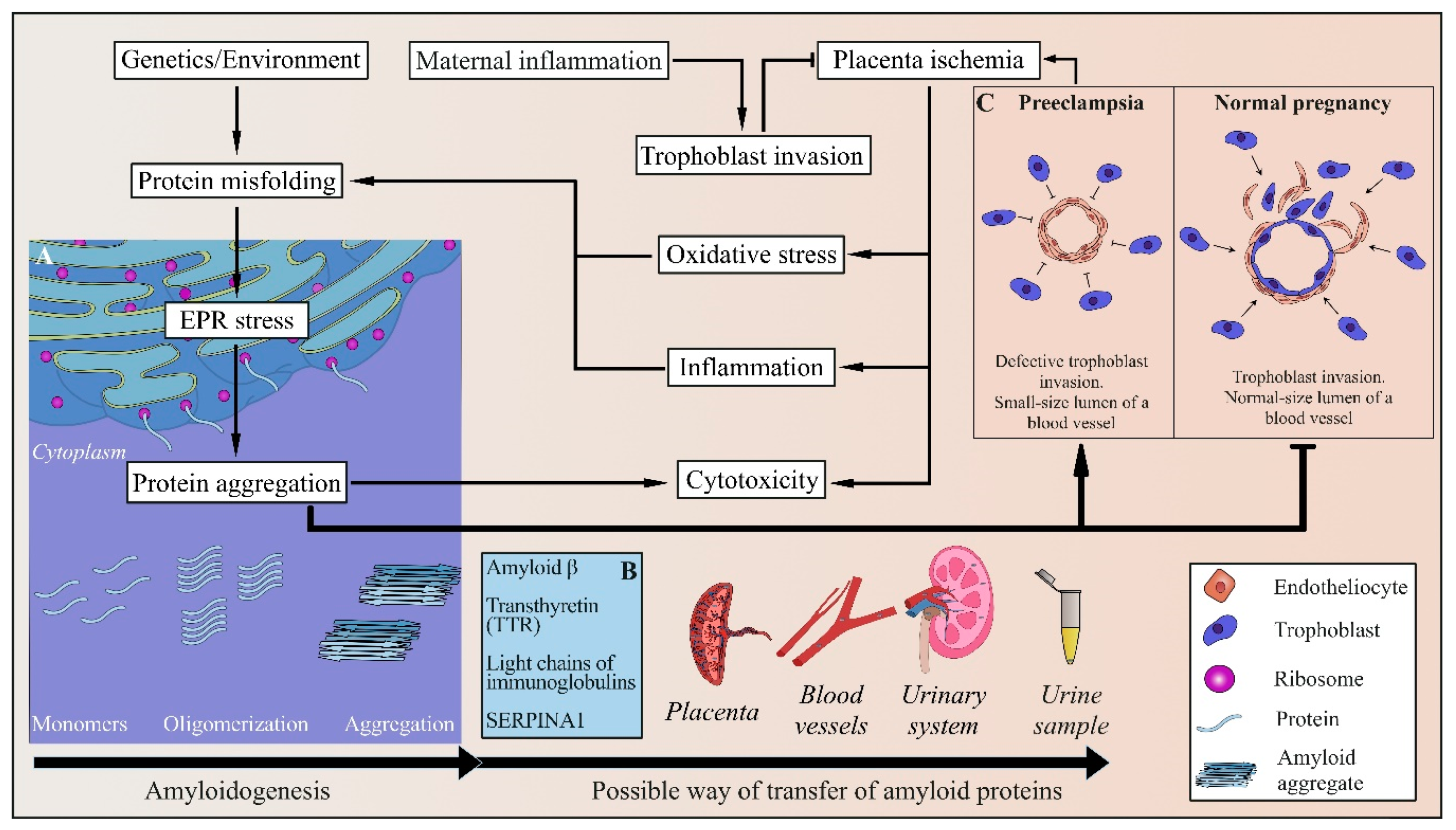{kind=link}
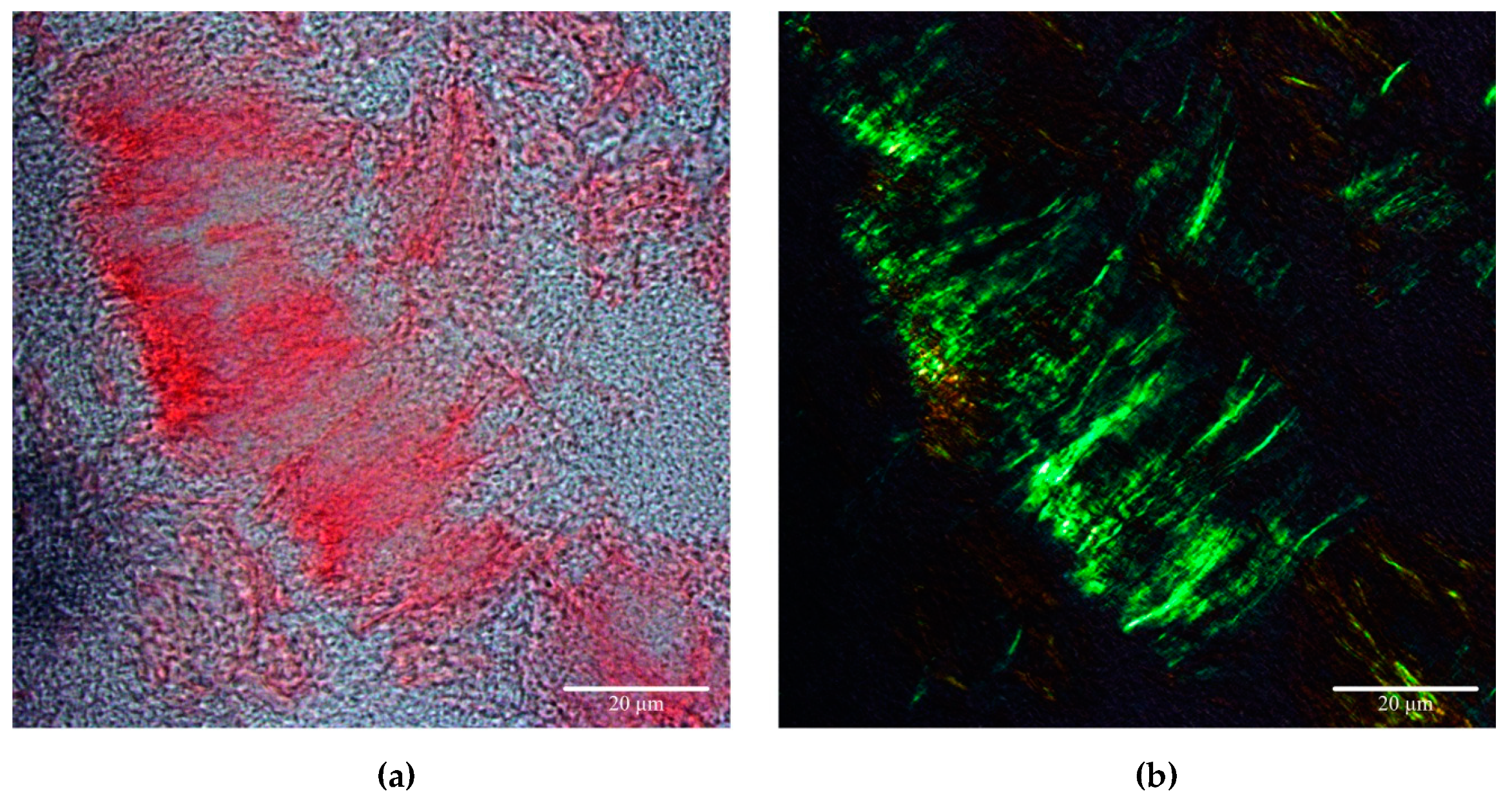{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Diagnosis of PE
3. Etiology and Pathogenesis of PE
4. Protein Misfolding and Amyloid Aggregation in PE
4.1. Amyloids and Amyloidogenic Diseases
4.2. Amyloids in PE
4.3. Alpha-1 Antitrypsin in PE
4.4. Light Chains of Immunoglobulins in PE
4.5. Amyloid β in PE
4.6. Transthyretin in PE
4.7. Possible Role of the Human Pregnancy Zone Protein
5. New Approaches to PE Diagnostics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PE | Preeclampsia |
| CR | Congo Red |
| CRD | Congo Red |
| BP | Blood Pressure |
| sFlt-1 | Soluble Fms-like tyrosine kinase-1 |
| sEng | soluble Endoglin |
| PLGF | Placental Growth Factor |
| sVEGFR | Vascular Endothelial Growth Factor |
| VEGF | Vascular Endothelial Growth Factor |
| ROS | Reactive Oxygen Species |
| HO | Heme Oxygenase |
| mRNA | messenger Ribonucleic Acid |
| NkB | Neurokinin B |
| AT1-AA | Autoantibodies to Angiotensin II receptor 1 |
| Apo E | Apolipoprotein E |
| TSEs | Transmissible Spongiform Encephalopathies |
| Aβ | Amyloid β peptide |
| EM | Electron Microscopy |
| ER | Endoplasmic Reticulum |
| TTR | Transthyretin |
| MS | Mass Spectrometry |
| igG | immunoglobulins |
| IFI-6 | Interferon-inducible protein 6-16 |
| APP | Amyloid Precursor Protein |
| sAPPa | soluble N-terminal fragment of APP |
| α2M | alpha-2-macroglobulin |
| PZP | Pregnancy Zone Protein |
| ThT | Thioflavin-T |
References
- Duley, L. The Global Impact of Pre-eclampsia and Eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef] [PubMed]
- American College of Obstetricians; Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2014, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sircar, M.; Thadhani, R.; Karumanchi, S.A. Pathogenesis of preeclampsia. Curr. Opin. Nephrol. Hypertens. 2015, 24, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.; Sibai, B. Maternal Mortality from Preeclampsia/Eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular Mechanisms of Preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5, a023473. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; van den Berg, C.; Alfirevic, Z.; O’Brien, S.; Röthlisberger, M.; Baker, P.; Kenny, L.; Kublickiene, K.; Duvekot, J. Early Pregnancy Biomarkers in Pre-Eclampsia: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2015, 16, 23035–23056. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.; Tan, M.Y.; O’Gorman, N.; Poon, L.C.; Syngelaki, A.; Wright, A.; Nicolaides, K.H. Predictive performance of the competing risk model in screening for preeclampsia. Am. J. Obstet. Gynecol. 2019, 220, e1–e199. [Google Scholar] [CrossRef] [Green Version]
- Schiettecatte, J.; Russcher, H.; Anckaert, E.; Mees, M.; Leeser, B.; Tirelli, A.S.; Fiedler, G.M.; Luthe, H.; Denk, B.; Smitz, J. Multicenter evaluation of the first automated Elecsys sFlt-1 and PlGF assays in normal pregnancies and preeclampsia. Clin. Biochem. 2010, 43, 768–770. [Google Scholar] [CrossRef]
- Salahuddin, S.; Lee, Y.; Vadnais, M.; Sachs, B.P.; Karumanchi, S.A.; Lim, K.-H. Diagnostic utility of soluble fms-like tyrosine kinase 1 and soluble endoglin in hypertensive diseases of pregnancy. Am. J. Obstet. Gynecol. 2007, 197, e1–e28. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.-H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sovio, U.; Gaccioli, F.; Cook, E.; Hund, M.; Charnock-Jones, D.S.; Smith, G.C.S. Prediction of Preeclampsia Using the Soluble fms-Like Tyrosine Kinase 1 to Placental Growth Factor Ratio: A Prospective Cohort Study of Unselected Nulliparous Women. Hypertension 2017, 69, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Wortelboer, E.; Koster, M.; Cuckle, H.; Stoutenbeek, P.; Schielen, P.; Visser, G. First-trimester placental protein 13 and placental growth factor: Markers for identification of women destined to develop early-onset pre-eclampsia. Int. J. Obstet. Gynaecol. 2010, 117, 1384–1389. [Google Scholar] [CrossRef]
- Nicolaides, K.H.; Bindra, R.; Turan, O.M.; Chefetz, I.; Sammar, M.; Meiri, H.; Tal, J.; Cuckle, H.S. A novel approach to first-trimester screening for early pre-eclampsia combining serum PP-13 and Doppler ultrasound. Ultrasound Obstet. Gynecol. 2005, 27, 13–17. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.-H.; Yuan, H.-T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Foidart, J.-M.; Munaut, C.; Chantraine, F.; Akolekar, R.; Nicolaides, K.H. Maternal plasma soluble endoglin at 11–13 weeks’ gestation in pre-eclampsia. Ultrasound Obstet. Gynecol. 2010, 35, 680–687. [Google Scholar] [CrossRef]
- Spencer, K.; Cowans, N.J.; Nicolaides, K.H. Low levels of maternal serum PAPP-A in the first trimester and the risk of pre-eclampsia. Prenat. Diagn. 2008, 28, 7–10. [Google Scholar] [CrossRef]
- Poon, L.C.; Nicolaides, K.H. Early Prediction of Preeclampsia. Obstet. Gynecol. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Diagnosis and management of preeclampsia and eclampsia. Int. J. Gynecol. Obstet. 2002, 77, 67–75. [CrossRef]
- Lowe, S.A.; Brown, M.A.; Dekker, G.A.; Gatt, S.; McLintock, C.K.; McMahon, L.P.; Mangos, G.; Moore, M.P.; Muller, P.; Paech, M.; et al. Guidelines for the management of hypertensive disorders of pregnancy 2008. Aust. N. Z. J. Obstet. Gynaecol. 2009, 49, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Thurnau, G.R. Pregnancy-induced hypertension without proteinuria: Is it true preeclampsia? South. Med. J. 1988, 81, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.L.; Mercer, B.M.; Friedman, S.A.; Sibai, B.M. Urinary dipstick protein: A poor predictor of absent or severe proteinuria. Am. J. Obstet. Gynecol. 1994, 170, 137–141. [Google Scholar] [CrossRef]
- Lindheimer, M.D.; Kanter, D. Interpreting abnormal proteinuria in pregnancy: The need for a more pathophysiological approach. Obstet. Gynecol. 2010, 115, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Buddle, M.L. Inadequacy of dipstick proteinuria in hypertensive pregnancy. Aust. N. Z. J. Obstet. Gynaecol. 1995, 35, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Grauer, G.F. Proteinuria: Measurement and interpretation. Top. Companion Anim. Med. 2011, 26, 121–127. [Google Scholar] [CrossRef]
- Shamshirsaz, A.A.; Paidas, M.; Krikun, G. Preeclampsia, Hypoxia, Thrombosis, and Inflammation. J. Pregnancy 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Roberts, J.M.; Gammill, H.S. Preeclampsia: Recent insights. Hypertension 2005, 46, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of Preeclampsia. Hypertens. Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef]
- Ødegård, R.A.; Vatten, L.J.; Nilsen, S.T.; Salvesen, K.; Austgulen, R. Risk factors and clinical manifestations of pre-eclampsia. Br. J. Obstet. Gynaecol. 2000, 107, 1410–1416. [Google Scholar] [CrossRef]
- Masuyama, H.; Segawa, T.; Sumida, Y.; Masumoto, A.; Inoue, S.; Akahori, Y.; Hiramatsu, Y. Different profiles of circulating angiogenic factors and adipocytokines between early- and late-onset pre-eclampsia. Int. J. Obstet. Gynaecol. 2010, 117, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in pathogenesis, definitions, and guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibai, B.M. Evaluation and management of severe preeclampsia before 34 weeks’ gestation. Am. J. Obstet. Gynecol. 2011, 205, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Leavey, K.; Bainbridge, S.A. Cox Large scale aggregate microarray analysis reveals three distinct molecular subclasses of human preeclampsia. PLoS ONE 2015, 10, e0116508. [Google Scholar] [CrossRef]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension Produced by Reduced Uterine Perfusion in Pregnant Rats Is Associated With Increased Soluble Fms-Like Tyrosine Kinase-1 Expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brosens, I.; Renaer, M. On the pathogenesis of placental infarcts in pre-eclampsia. Int. J. Obstet. Gynaecol. 1972, 79, 794–799. [Google Scholar] [CrossRef]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble Endoglin and Other Circulating Antiangiogenic Factors in Preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Harihana, N.; Shoemaker, A.; Wagner, S. Pathophysiology of hypertension in preeclampsia. Clin. Pr. 2016, 13, 33–37. [Google Scholar]
- Than, N.G.; Romero, R.; Tarca, A.L.; Kekesi, K.A.; Xu, Y.; Xu, Z.; Juhasz, K.; Bhatti, G.; Leavitt, R.J.; Gelencser, Z.; et al. Integrated Systems Biology Approach Identifies Novel Maternal and Placental Pathways of Preeclampsia. Front. Immunol. 2018, 9, 1661. [Google Scholar] [CrossRef]
- El-Sayed, A.A.F. Preeclampsia: A review of the pathogenesis and possible management strategies based on its pathophysiological derangements. Taiwan. J. Obstet. Gynecol. 2017, 56, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuchi, A.; Hirashima, C.; Suzuki, H.; Takahashi, K.; Yoshida, M.; Matsubara, S.; Suzuki, M. Evaluation of a new and automated electrochemiluminescence immunoassay for plasma sFlt-1 and PlGF levels in women with preeclampsia. Hypertens. Res. 2010, 33, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, U.B.; Kronborg, C.S.; von Dadelszen, P.; Kupfer, K.; Lee, S.-W.; Vittinghus, E.; Allen, J.G.; Redman, C.W. A single rapid point-of-care placental growth factor determination as an aid in the diagnosis of preeclampsia. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2012, 2, 8–15. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Myatt, L.; Cui, X. Oxidative stress in the placenta. Histochem. Cell Biol. 2004, 122, 369–382. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Spasic-Boskovic, O.; Jauniaux, E.; Charnock-Jones, D.S.; Burton, G.J. Nuclear Factor-κB, p38, and Stress-Activated Protein Kinase Mitogen-Activated Protein Kinase Signaling Pathways Regulate Proinflammatory Cytokines and Apoptosis in Human Placental Explants in Response to Oxidative Stress. Am. J. Pathol. 2007, 170, 1511–1520. [Google Scholar] [CrossRef]
- Zenclussen, A.C.; Lim, E.; Knoeller, S.; Knackstedt, M.; Hertwig, K.; Hagen, E.; Klapp, B.F.; Arck, P.C. Heme Oxygenases in Pregnancy II: HO-2 is Downregulated in Human Pathologic Pregnancies. Am. J. Reprod. Immunol. 2003, 50, 66–76. [Google Scholar] [CrossRef]
- Lyall, F.; Barber, A.; Myatt, L.; Bulmer, J.N.; Robson, S.C. Hemeoxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J. 2000, 14, 208–219. [Google Scholar] [CrossRef] [PubMed]
- George, E.M.; Granger, J.P. Heme oxygenase in pregnancy and preeclampsia. Curr. Opin. Nephrol. Hypertens. 2013, 22, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, S.; Farley, A.; McLaughlin, B.; Graham, C.; Marks, G.; Nakatsu, K.; Brien, J.; Smith, G. Carbon Monoxide Decreases Perfusion Pressure in Isolated Human Placenta. Placenta 2002, 23, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative Regulation of Soluble Flt-1 and Soluble Endoglin Release by Heme Oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaig, D.; Lyall, F. Inhibitors of Heme Oxygenase Reduce Invasion of Human Primary Cytotrophoblast Cells In vitro. Placenta 2009, 30, 536–538. [Google Scholar] [CrossRef]
- Costantine, M.M.; Tamayo, E.; Lu, F.; Bytautiene, E.; Longo, M.; Hankins, G.D.V.; Saade, G.R. Using Pravastatin to Improve the Vascular Reactivity in a Mouse Model of Soluble Fms-Like Tyrosine Kinase-1–Induced Preeclampsia. Obstet. Gynecol. 2010, 116, 114–120. [Google Scholar] [CrossRef]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Bujold, E.; Chaiworapongsa, T.; Gomez, R.; Yoon, B.H.; Thaler, H.T.; Rotmensch, S.; Romero, R. Failure of physiologic transformation of the spiral arteries in patients with preterm labor and intact membranes. Am. J. Obstet. Gynecol. 2003, 189, 1063–1069. [Google Scholar] [CrossRef]
- Dommisse, J.; Tiltman, A.J. Placental bed biopsies in placental abruption. Int. J. Obstet. Gynaecol. 1992, 99, 651–654. [Google Scholar] [CrossRef]
- Page, N.; Butlin, D.; Manyonda, I.; Lowry, P. The development of a genetic profile of placental gene expression during the first trimester of pregnancy: A potential tool for identifying novel secreted markers. Fetal Diagn. Ther. 2000, 15, 237–245. [Google Scholar] [CrossRef]
- Zulfikaroglu, E.; Ugur, M.; Taflan, S.; Ugurlu, N.; Atalay, A.; Kalyoncu, S. Neurokinin B levels in maternal and umbilical cord blood in preeclamptic and normal pregnancies. J. Perinat. Med. 2007, 35, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Page, N.M.; Kemp, C.F.; Butlin, D.J.; Lowry, P.J. Placental peptides as markers of gestational disease. Reproduction 2002, 123, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, C.; DiMarzo, L.; Morrish, D.W.; Kaufman, S. Neurokinin B causes concentration-dependent relaxation of isolated human placental resistance vessels. Regul. Pept. 2004, 117, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Wu, J.; Murray, J.K.; Gellman, S.H.; Wozniak, M.A.; Keely, P.J.; Boyer, M.E.; Gomez, T.M.; Hasso, S.M.; Fallon, J.F.; et al. An antiangiogenic neurokinin-B/thromboxane A2 regulatory axis. J. Cell Biol. 2006, 174, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Page, N.M. Neurokinin B and pre-eclampsia: A decade of discovery. Reprod. Biol. Endocrinol. 2010, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Hoang, V.M.; Foulk, R.; Clauser, K.; Burlingame, A.; Gibson, B.W.; Fisher, S.J. Functional Proteomics: Examining the Effects of Hypoxia on the Cytotrophoblast Protein Repertoire. Biochemistry 2001, 40, 4077–4086. [Google Scholar] [CrossRef]
- Sawicki, G.; Dakour, J.; Morrish, D.W. Functional proteomics of neurokinin B in the placenta indicates a novel role in regulating cytotrophoblast antioxidant defences. Proteomics 2003, 3, 2044–2051. [Google Scholar] [CrossRef]
- Xie, F.; von Dadelszen, P.; Nadeau, J. CMV infection, TLR-2 and -4 expression, and cytokine profiles in early-onset preeclampsia with HELLP syndrome. Am. J. Reprod. Immunol. 2014, 71, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Molvarec, A.; Szarka, A.; Walentin, S.; Bekő, G.; Karádi, I.; Prohászka, Z.; Rigó, J., Jr. Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod. Biol. Endocrinol. 2011, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jüpner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Investig. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Romero, R.; Yoshimatsu, J.; Espinoza, J.; Kim, Y.M.; Park, K.; Kalache, K.; Edwin, S.; Bujold, E.; Gomez, R. Soluble adhesion molecule profile in normal pregnancy and pre-eclampsia. J. Matern. Neonatal Med. 2002, 12, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Bretelle, F.; Sabatier, F.; Blann, A.; D’Ercole, C.; Boutière, B.; Mutin, M.; Boubli, L.; Sampol, J.; Dignat-George, F. Maternal endothelial soluble cell adhesion molecules with isolated small for gestational age fetuses: Comparison with pre-eclampsia. Br. J. Obstet. Gynaecol. 2001, 108, 1277–1282. [Google Scholar]
- Francoual, J.; Audibert, F.; Trioche, P.; Chalas, J.; Capel, L.; Lindenbaum, A.; Labrune, P.; Frydman, R. Erratum: Is a Polymorphism of the Apolipoprotein E Gene Associated with Preeclampsia? Hypertens. Pregnancy 2003, 21, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Belo, L.; Gaffney, D.; Caslake, M.; Santos-Silva, A.; Pereira-Leite, L.; Quintanilha, A.; Rebelo, I. Apolipoprotein E and cholesteryl ester transfer protein polymorphisms in normal and preeclamptic pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 112, 9–15. [Google Scholar] [CrossRef]
- Aguzzi, A.; Haass, C. Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science 2003, 302, 814–818. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Wiggins, R.C. Prions and the Transmissible Spongiform Encephalopathies. In Metabolic Encephalopathy; McCandless, D., Ed.; Springer: New York, NY, USA, 2009; pp. 531–550. [Google Scholar]
- Belay, E.D. Transmissible Spongiform Encephalopathies in Humans. Annu. Rev. Microbiol. 1999, 53, 283–314. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, M.D.; Legname, G. Transmissible spongiform encephalopathies. In Protein Misfolding in Neurodegenerative Diseases: Mechanisms and Therapeutic Strategies; CRC Press; Smith, H.J., Simons, C., Sewell, R.D.E., Eds.; Taylor and Francis Group: Abingdon, UK, 2007; pp. 221–248. [Google Scholar]
- Iwasaki, Y. Creutzfeldt-Jakob disease. Neuropathology. 2017, 2, 174–188. [Google Scholar] [CrossRef]
- Brown, K.; Mastrianni, J.A. The Prion Diseases. J. Geriatr. Psychiatry Neurol. 2010, 23, 277–298. [Google Scholar] [CrossRef]
- Bhadbhade, A.; Cheng, D.W. Amyloid Precursor Protein Processing in Alzheimer’s Disease. Iran. J. Child Neurol. 2012, 34, 185–204. [Google Scholar]
- Iqbal, K.; Alonso, A.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. α-Synuclein and neuronal cell death. Mol. Neurobiol. 2013, 47, 466–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Wang, Z.; Lei, H.; Zhang, W.; Duan, Y. Dual binding modes of Congo red to amyloid protofibril surface observed in molecular dynamics simulations. J. Am. Chem. Soc. 2007, 129, 1225–1232. [Google Scholar] [CrossRef]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- Ivancic, V.A.; Ekanayake, O.; Lazo, N.D. Binding Modes of Thioflavin T on the Surface of Amyloid Fibrils Studied by NMR. ChemPhysChem 2016, 17, 2461–2464. [Google Scholar] [CrossRef]
- Levine, H. Thioflavine t interaction with amyloid βsheet structures. Amyloid 1995, 2, 1–6. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β (1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and x-ray diffraction. Adv. Protein Chem. 1997, 50, 123–159. [Google Scholar] [PubMed]
- Chandramowlishwaran, P.; Sun, M.; Casey, K.L.; Romanyuk, A.V.; Grizel, A.V.; Sopova, J.V.; Rubel, A.A.; Nussbaum-Krammer, C.; Vorberg, I.M.; Chernoff, Y.O. Mammalian amyloidogenic proteins promote prion nucleation in yeast. J. Biol. Chem. 2018, 293, 3436–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubel, A.A.; Ryzhova, T.A.; Antonets, K.S.; Chernoff, Y.O.; Galkin, A.P. Identification of PrP sequences essential for the interaction between the PrP polymers and Aβ peptide in a yeast-based assay. Prion 2013, 7, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Buhimschi, I.A.; Zhao, G.; Funai, E.F.; Harris, N.; Sasson, I.E.; Bernstein, I.M.; Saade, G.R.; Buhimschi, C.S. Proteomic profiling of urine identifies specific fragments of SERPINA1 and albumin as biomarkers of preeclampsia. Am. J. Obstet. Gynecol. 2008, 199, e1–e551. [Google Scholar] [CrossRef]
- Millen, K.R.; Buhimschi, C.S.; Zhao, G.; Rood, K.M.; Tabbah, S.; Buhimschi, I.A. Serum and Urine Thioflavin-T-Enhanced Fluorescence in Severe Preeclampsia. Hypertension 2018, 71, 1185–1192. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra92. [Google Scholar] [CrossRef]
- Tong, M.; Cheng, S.; Chen, Q.; DeSousa, J.; Stone, P.R.; James, J.L.; Chamley, L.W.; Sharma, S. Aggregated transthyretin is specifically packaged into placental nano-vesicles in preeclampsia. Sci. Rep. 2017, 7, 6694. [Google Scholar] [CrossRef] [Green Version]
- Cater, J.H.; Kumita, J.R.; Zeineddine Abdallah, R.; Zhao, G.; Bernardo-Gancedo, A.; Henry, A.; Winata, W.; Chi, M.; Grenyer, B.S.F.; Townsend, M.L.; et al. Human pregnancy zone protein stabilizes misfolded proteins including preeclampsia- and Alzheimer’s-associated amyloid beta peptide. Proc. Natl. Acad. Sci. USA 2019, 116, 6101–6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-B.; Nakashima, A.; Sharma, S. Understanding Pre-Eclampsia Using Alzheimer’s Etiology: An Intriguing Viewpoint. Am. J. Reprod. Immunol. 2016, 75, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, G.G.; Pechmann, S.; Dobson, C.M.; Vendruscolo, M. Life on the edge: A link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 2007, 32, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.R.; Crowder, C.M. Protein Misfolding Induces Hypoxic Preconditioning via a Subset of the Unfolded Protein Response Machinery. Mol. Cell. Biol. 2010, 30, 5033–5042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschen, W.; Mengesdorf, T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 2005, 38, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Jian, B.; Hsieh, C.H.; Chen, J.; Choudhry, M.; Bland, K.; Chaudry, I.; Raju, R. Activation of endoplasmic reticulum stress response following trauma-hemorrhage. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [Green Version]
- Yoshiike, Y.; Kayed, R.; Milton, S.C.; Takashima, A.; Glabe, C.G. Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromol. Med. 2007, 9, 270–275. [Google Scholar] [CrossRef]
- Shirahama, T. High-resolution electron microscopic analysis of the amyloid fibril. J. Cell Biol. 1967, 33, 679–708. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Katzmann, J.A.; Larson, D.R.; Colby, C.L.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Leung, N.; Zeldenrust, S.R.; et al. Serum immunoglobulin free light-chain measurement in primary amyloidosis: Prognostic value and correlations with clinical features. Blood 2010, 116, 5126–5129. [Google Scholar] [CrossRef]
- Huntington, J.A. Serpin structure, function and dysfunction. J. Thromb. Haemost. 2011, 9, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Engström, G.; Janzon, L.; Berglund, G.; Lind, P.; Stavenow, L.; Hedblad, B.; Lindgärde, F. Blood Pressure Increase and Incidence of Hypertension in Relation to Inflammation-Sensitive Plasma Proteins. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2054–2058. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.R.; Wong, P.S.; Travis, J. Enzymatic inactivation of human alpha-1-proteinase inhibitor by neutrophil myeloperoxidase. Biochem. Biophys. Res. Commun. 1979, 88, 402–409. [Google Scholar] [CrossRef]
- Lomas, D.A. Polymerisation underlies alpha1-antitrypsin deficiency, dementia and other serpinopathies. Front. Biosci. 2004, 9, 2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Alvarado, M. Amyloid Formation in Light Chain Amyloidosis. Curr. Top. Med. Chem. 2013, 12, 2523–2533. [Google Scholar] [CrossRef]
- Bellotti, V.; Mangione, P.; Merlini, G. Review: Immunoglobulin Light Chain Amyloidosis—The Archetype of Structural and Pathogenic Variability. J. Struct. Biol. 2000, 130, 280–289. [Google Scholar] [CrossRef]
- Benson, M.D.; Liepnieks, J.J.; Kluve-Beckerman, B. Hereditary systemic immunoglobulin light-chain amyloidosis. Blood 2015, 125, 3281–3286. [Google Scholar] [CrossRef] [Green Version]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Buxbaum, J.N. The systemic amyloidoses. Curr. Opin. Rheumatol. 2004, 16, 67–75. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid β peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 1993, 75, 1039–1042. [Google Scholar] [CrossRef]
- Clarris, H.J.; Key, B.; Beyreuther, K.; Masters, C.L.; Small, D.H. Expression of the amyloid protein precursor of Alzheimer’s disease in the developing rat olfactory system. Brain Res. Dev. Brain Res. 1995, 88, 87–95. [Google Scholar] [CrossRef]
- Muresan, V.; Varvel, N.H.; Lamb, B.T.; Muresan, Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J. Neurosci. 2009, 29, 3565–3578. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease Beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.-G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Planté-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanetj. Rare Dis. 2013, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Kalkunte, S.S.; Neubeck, S.; Norris, W.E.; Cheng, S.-B.; Kostadinov, S.; Vu Hoang, D.; Ahmed, A.; von Eggeling, F.; Shaikh, Z.; Padbury, J.; et al. Transthyretin Is Dysregulated in Preeclampsia, and Its Native Form Prevents the Onset of Disease in a Preclinical Mouse Model. Am. J. Pathol. 2013, 183, 1425–1436. [Google Scholar] [CrossRef] [Green Version]
- Kalkunte, S.; Boij, R.; Norris, W.; Friedman, J.; Lai, Z.; Kurtis, J.; Lim, K.-H.; Padbury, J.F.; Matthiesen, L.; Sharma, S. Sera from Preeclampsia Patients Elicit Symptoms of Human Disease in Mice and Provide a Basis for an in Vitro Predictive Assay. Am. J. Pathol. 2010, 177, 2387–2398. [Google Scholar] [CrossRef]
- Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular Chaperones and Proteostasis. Annu. Rev. Biochem. 2013, 82, 295–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin Has Chaperone-like Activity Similar to That of Small Heat Shock Proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Kumita, J.R.; Meehan, S.; Dobson, C.M.; Wilson, M.R. α2-Macroglobulin and Haptoglobin Suppress Amyloid Formation by Interacting with Prefibrillar Protein Species. J. Biol. Chem. 2009, 284, 4246–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blacker, D.; Wilcox, M.A.; Laird, N.M.; Rodes, L.; Horvath, S.M.; Go, R.C.P.; Perry, R.; Watson, B.; Bassett, S.S.; McInnis, M.G.; et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat. Genet. 1998, 19, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Ekelund, L.; Laurell, C.B. The pregnancy zone protein response during gestation: A metabolic challenge. Scand. J. Clin. Lab. Investig. 1994, 54, 623–629. [Google Scholar] [CrossRef]
- Tatarinov, I.S.; Mesniankina, N.V.; Nikulina, D.M. Immunochemical indentification of beta globulin of the “pregnancy zone” in the blood serum of patients with hydatid mole and chorioepithelioma]. Akush. Ginekol. 1974, 5, 67–68. [Google Scholar]
- Tatarinov, I.S.; Masiukevich, V.N.; Mesniankina, N.V.; Parfenova, L.F. Immunochemical identification of a new alpha 2 globulin in the blood serum of pregnant women]. Akush. Ginekol. 1970, 46, 25–28. [Google Scholar]
- Perni, U.; Sison, C.; Sharma, V.; Helseth, G.; Hawfield, A.; Suthanthiran, M.; August, P. Angiogenic Factors in Superimposed Preeclampsia. Hypertension 2012, 59, 740–746. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Cerdeira, A.S.; Wenger, J.; Salahuddin, S.; Lim, K.-H.; Ralston, S.J.; Thadhani, R.I.; Karumanchi, S.A. Plasma Concentrations of Soluble Endoglin versus Standard Evaluation in Patients with Suspected Preeclampsia. PLoS ONE 2012, 7, e48259. [Google Scholar] [CrossRef]
- Akolekar, R.; Syngelaki, A.; Poon, L.; Wright, D.; Nicolaides, K.H. Competing risks model in early screening for preeclampsia by biophysical and biochemical markers. Fetal Diagn. Ther. 2013, 33, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeeva, V.A.; Zakharova, N.V.; Bugrova, A.E.; Starodubtseva, N.L.; Indeykina, M.I.; Kononikhin, A.S.; Frankevich, V.E.; Nikolaev, E.N. The high-resolution mass spectrometry study of the protein composition of amyloid-like urine aggregates associated with preeclampsia. Eur. J. Mass Spectrom. 2019, 146906671986007. [Google Scholar] [CrossRef] [PubMed]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39, BSR20181415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halimi, M.; Dayan-Amouyal, Y.; Kariv-Inbal, Z.; Friedman-Levi, Y.; Mayer-Sonnenfeld, T.; Gabizon, R. Prion urine comprises a glycosaminoglycan-light chain IgG complex that can be stained by Congo red. J. Virol. Methods 2006, 133, 205–210. [Google Scholar] [CrossRef]
- Rood, K.M.; Buhimschi, C.S.; Dible, T.; Webster, S.; Zhao, G.; Samuels, P.; Buhimschi, I.A. Congo Red Dot Paper Test for Antenatal Triage and Rapid Identification of Preeclampsia. EClinicalMedicine 2019, 8, 47–56. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerasimova, E.M.; Fedotov, S.A.; Kachkin, D.V.; Vashukova, E.S.; Glotov, A.S.; Chernoff, Y.O.; Rubel, A.A. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. Int. J. Mol. Sci. 2019, 20, 6183. https://doi.org/10.3390/ijms20246183
Gerasimova EM, Fedotov SA, Kachkin DV, Vashukova ES, Glotov AS, Chernoff YO, Rubel AA. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. International Journal of Molecular Sciences. 2019; 20(24):6183. https://doi.org/10.3390/ijms20246183
Chicago/Turabian StyleGerasimova, Elizaveta M., Sergey A. Fedotov, Daniel V. Kachkin, Elena S. Vashukova, Andrey S. Glotov, Yury O. Chernoff, and Aleksandr A. Rubel. 2019. "Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics" International Journal of Molecular Sciences 20, no. 24: 6183. https://doi.org/10.3390/ijms20246183
APA StyleGerasimova, E. M., Fedotov, S. A., Kachkin, D. V., Vashukova, E. S., Glotov, A. S., Chernoff, Y. O., & Rubel, A. A. (2019). Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. International Journal of Molecular Sciences, 20(24), 6183. https://doi.org/10.3390/ijms20246183