The Role of IgA in the Pathogenesis of IgA Nephropathy


Abstract
:1. Introduction
2. The Biological Characteristics of IgA in Humans
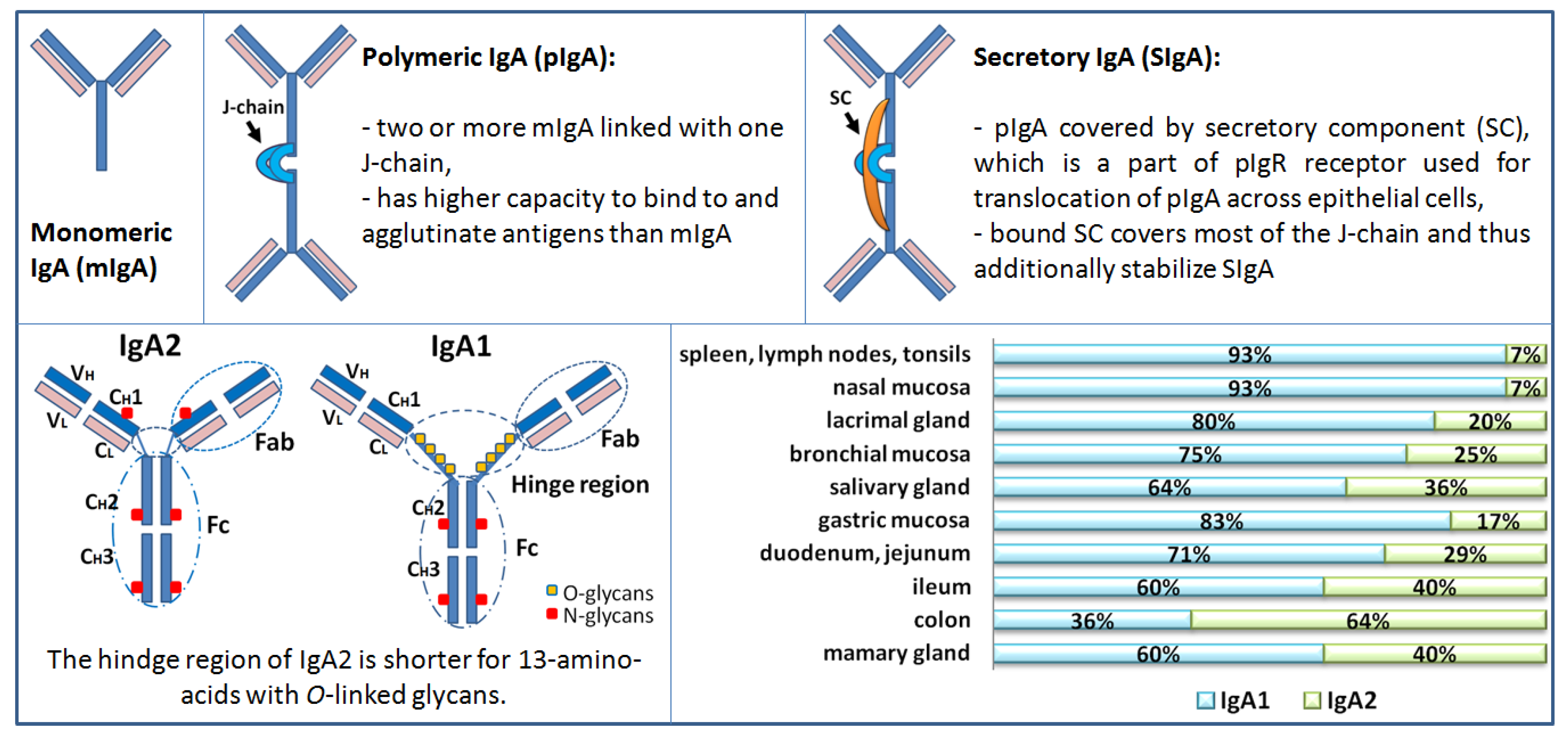
3. Structure of Human IgA
4. IgA Clearance and IgA Receptors
5. IgA Deposits in Kidney
6. Proposed Risk Factors in the Pathogenesis of IgAN
6.1. Increased Production of Galactose-Deficient IgA1 (Gd-IgA1)
6.2. Genetic Factors Associated with Gd-IgA1 or IgAN
6.3. Nongenetic Factors that Modify Glycosylation of IgA1
7. Decreased IgA Clearance and IgA Receptor Alterations
8. Four Hit Hypothesis and Other Hypotheses
The Role of SIgA and Complement in IgAN
9. Biomarkers of IgAN
Author Contributions
Funding
Conflicts of Interest
References
- Schena, F.P.; Nistor, I. Epidemiology of IgA Nephropathy: A Global Perspective. Semin. Nephrol. 2018, 38, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S. Pathology of IgA nephropathy. Nat. Rev. Nephrol. 2014, 10, 445–454. [Google Scholar] [CrossRef] [PubMed]
- McGrogan, A.; Franssen, C.F.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionaki, S.; Panagiotellis, K.; Melexopoulou, C.; Boletis, J.N. The clinical course of IgA nephropathy after kidney transplantation and its management. Transplant. Rev. (Orlando) 2017, 31, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wyld, M.L.; Chadban, S.J. Recurrent IgA Nephropathy After Kidney Transplantation. Transplantation 2016, 100, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Sofue, T.; Suzuki, H.; Ueda, N.; Kushida, Y.; Minamino, T. Post-transplant immunoglobulin A deposition and nephropathy in allografts. Nephrology (Carlton) 2018, 23 (Suppl. 2), 4–9. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, L.; Robert, T.; Vuiblet, V.; Tabary, T.; Braconnier, A.; Dramé, M.; Toupance, O.; Rieu, P.; Monteiro, R.C.; Touré, F. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015, 88, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Garnier, A.S.; Duveau, A.; Demiselle, J.; Croué, A.; Subra, J.F.; Sayegh, J.; Augusto, J.F. Early post-transplant serum IgA level is associated with IgA nephropathy recurrence after kidney transplantation. PLoS ONE 2018, 13, e0196101. [Google Scholar] [CrossRef]
- Berthoux, F.; Suzuki, H.; Mohey, H.; Maillard, N.; Mariat, C.; Novak, J.; Julian, B.A. Prognostic Value of Serum Biomarkers of Autoimmunity for Recurrence of IgA Nephropathy after Kidney Transplantation. J. Am. Soc. Nephrol. 2017, 28, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, T.; Nitta, K.; Suzuki, K.; Honda, K.; Horita, S.; Uchida, K.; Yumura, W.; Tanabe, K.; Toma, H.; Nihei, H.; et al. Latent IgA deposition from donor kidney is the major risk factor for recurrent IgA nephropathy in renal transplantation. Clin. Transplant. 2005, 19 (Suppl. 14), 41–48. [Google Scholar] [CrossRef]
- Moroni, G.; Belingheri, M.; Frontini, G.; Tamborini, F.; Messa, P. Immunoglobulin A Nephropathy. Recurrence After Renal Transplantation. Front. Immunol. 2019, 10, 1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conley, M.E.; Delacroix, D.L. Intravascular and mucosal immunoglobulin A: two separate but related systems of immune defense? Ann. Intern. Med. 1987, 106, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Secretory IgA: Designed for Anti-Microbial Defense. Front. Immunol. 2013, 4, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakkanen, S.H.; Kantele, J.M.; Moldoveanu, Z.; Hedges, S.; Häkkinen, M.; Mestecky, J.; Kantele, A. Expression of homing receptors on IgA1 and IgA2 plasmablasts in blood reflects differential distribution of IgA1 and IgA2 in various body fluids. Clin. Vaccine Immunol. 2010, 17, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.; Moldoveanu, Z.; Renfrow, M.B.; Yanagihara, T.; Suzuki, H.; Raska, M.; Hall, S.; Brown, R.; Huang, W.Q.; Goepfert, A.; et al. IgA nephropathy and Henoch-Schoenlein purpura nephritis: aberrant glycosylation of IgA1, formation of IgA1-containing immune complexes, and activation of mesangial cells. Contrib. Nephrol. 2007, 157, 134–138. [Google Scholar] [CrossRef]
- Novak, J.; Tomana, M.; Kilian, M.; Coward, L.; Kulhavy, R.; Barnes, S.; Mestecky, J. Heterogeneity of O-glycosylation in the hinge region of human IgA1. Mol. Immunol. 2000, 37, 1047–1056. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Gate-keeper function of the intestinal epithelium. Benef. Microbes 2013, 4, 67–82. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Secretory immunity with special reference to the oral cavity. J. Oral Microbiol. 2013, 5. [Google Scholar] [CrossRef]
- Stuchlova Horynova, M.; Vrablikova, A.; Stewart, T.J.; Takahashi, K.; Czernekova, L.; Yamada, K.; Suzuki, H.; Julian, B.A.; Renfrow, M.B.; Novak, J.; et al. N-acetylgalactosaminide α2,6-sialyltransferase II is a candidate enzyme for sialylation of galactose-deficient IgA1, the key autoantigen in IgA nephropathy. Nephrol. Dial. Transplant. 2015, 30, 234–238. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.W.; Ding, J.L. The unexplored roles of human serum IgA. DNA Cell Biol. 2014, 33, 823–829. [Google Scholar] [CrossRef]
- Moldoveanu, Z.; Moro, I.; Radl, J.; Thorpe, S.R.; Komiyama, K.; Mestecky, J. Site of catabolism of autologous and heterologous IgA in non-human primates. Scand J. Immunol. 1990, 32, 577–583. [Google Scholar] [CrossRef]
- Weigel, P.H.; Yik, J.H. Glycans as endocytosis signals: the cases of the asialoglycoprotein and hyaluronan/chondroitin sulfate receptors. Biochim. Biophys. Acta 2002, 1572, 341–363. [Google Scholar] [CrossRef]
- Moura, I.C.; Centelles, M.N.; Arcos-Fajardo, M.; Malheiros, D.M.; Collawn, J.F.; Cooper, M.D.; Monteiro, R.C. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 2001, 194, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, K.; Wimbury, D.; Pawluczyk, I.; Muto, M.; Bhachu, J.; Mertens, P.R.; Feehally, J.; Barratt, J. β1,4-galactosyltransferase 1 is a novel receptor for IgA in human mesangial cells. Kidney Int. 2017, 92, 1458–1468. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, A.; Sakamoto, N.; Shimizu, Y.; Shibuya, K.; Osawa, M.; Hiroyama, T.; Eyre, H.J.; Sutherland, G.R.; Endo, Y.; Fujita, T.; et al. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat. Immunol. 2000, 1, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Lamkhioued, B.; Gounni, A.S.; Gruart, V.; Pierce, A.; Capron, A.; Capron, M. Human eosinophils express a receptor for secretory component. Role in secretory IgA-dependent activation. Eur. J. Immunol. 1995, 25, 117–125. [Google Scholar] [CrossRef]
- Mantis, N.J.; Cheung, M.C.; Chintalacharuvu, K.R.; Rey, J.; Corthésy, B.; Neutra, M.R. Selective adherence of IgA to murine Peyer’s patch M cells: evidence for a novel IgA receptor. J. Immunol. 2002, 169, 1844–1851. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.J.; Fuchs, A.; Colonna, M. Cutting edge: human FcRL4 and FcRL5 are receptors for IgA and IgG. J. Immunol. 2012, 188, 4741–4745. [Google Scholar] [CrossRef] [Green Version]
- Mostov, K.E.; Friedlander, M.; Blobel, G. The receptor for transepithelial transport of IgA and IgM contains multiple immunoglobulin-like domains. Nature 1984, 308, 37–43. [Google Scholar] [CrossRef]
- Baumann, J.; Park, C.G.; Mantis, N.J. Recognition of secretory IgA by DC-SIGN: implications for immune surveillance in the intestine. Immunol. Lett. 2010, 131, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochereau, N.; Drocourt, D.; Perouzel, E.; Pavot, V.; Redelinghuys, P.; Brown, G.D.; Tiraby, G.; Roblin, X.; Verrier, B.; Genin, C.; et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013, 11, e1001658. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.L.; van den Berg, C.W.; Bowen, D.J. ASGR1 and ASGR2, the Genes that Encode the Asialoglycoprotein Receptor (Ashwell Receptor), Are Expressed in Peripheral Blood Monocytes and Show Interindividual Differences in Transcript Profile. Mol. Biol. Int. 2012, 2012, 283974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Hu, B.; Yang, Y.; Ma, Z.; Yu, Y.; Liu, S.; Wang, B.; Zhao, X.; Lu, M.; Yang, D. A new splice variant of the major subunit of human asialoglycoprotein receptor encodes a secreted form in hepatocytes. PLoS ONE 2010, 5, e12934. [Google Scholar] [CrossRef] [PubMed]
- Motegi, Y.; Kita, H.; Kato, M.; Morikawa, A. Role of secretory IgA, secretory component, and eosinophils in mucosal inflammation. Int. Arch. Allergy Immunol. 2000, 122 (Suppl. 1), 25–27. [Google Scholar] [CrossRef]
- Mota, G.; Manciulea, M.; Cosma, E.; Popescu, I.; Hirt, M.; Jensen-Jarolim, E.; Calugaru, A.; Galatiuc, C.; Regalia, T.; Tamandl, D.; et al. Human NK cells express Fc receptors for IgA which mediate signal transduction and target cell killing. Eur. J. Immunol. 2003, 33, 2197–2205. [Google Scholar] [CrossRef]
- Aleyd, E.; Heineke, M.H.; van Egmond, M. The era of the immunoglobulin A Fc receptor FcαRI; its function and potential as target in disease. Immunol. Rev. 2015, 268, 123–138. [Google Scholar] [CrossRef]
- Suzuki, K.; Honda, K.; Tanabe, K.; Toma, H.; Nihei, H.; Yamaguchi, Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003, 63, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Varis, J.; Rantala, I.; Pasternack, A.; Oksa, H.; Jäntti, M.; Paunu, E.S.; Pirhonen, R. Immunoglobulin and complement deposition in glomeruli of 756 subjects who had committed suicide or met with a violent death. J. Clin. Pathol. 1993, 46, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Sinniah, R. Occurrence of mesangial IgA and IgM deposits in a control necropsy population. J. Clin. Pathol. 1983, 36, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Waldherr, R.; Rambausek, M.; Duncker, W.D.; Ritz, E. Frequency of mesangial IgA deposits in a non-selected autopsy series. Nephrol. Dial. Transplant. 1989, 4, 943–946. [Google Scholar] [CrossRef]
- Iwasaki, H.; Zhang, Y.; Tachibana, K.; Gotoh, M.; Kikuchi, N.; Kwon, Y.D.; Togayachi, A.; Kudo, T.; Kubota, T.; Narimatsu, H. Initiation of O-glycan synthesis in IgA1 hinge region is determined by a single enzyme, UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 2. J. Biol. Chem. 2003, 278, 5613–5621. [Google Scholar] [CrossRef] [Green Version]
- Wandall, H.H.; Irazoqui, F.; Tarp, M.A.; Bennett, E.P.; Mandel, U.; Takeuchi, H.; Kato, K.; Irimura, T.; Suryanarayanan, G.; Hollingsworth, M.A.; et al. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology 2007, 17, 374–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Tachibana, K.; Zhang, Y.; Iwasaki, H.; Kameyama, A.; Cheng, L.; Guo, J.; Hiruma, T.; Togayachi, A.; Kudo, T.; et al. Cloning and characterization of a novel UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase, pp-GalNAc-T14. Biochem. Biophys. Res. Commun. 2003, 300, 738–744. [Google Scholar] [CrossRef]
- Gale, D.P.; Molyneux, K.; Wimbury, D.; Higgins, P.; Levine, A.P.; Caplin, B.; Ferlin, A.; Yin, P.; Nelson, C.P.; Stanescu, H.; et al. Galactosylation of IgA1 Is Associated with Common Variation in. J. Am. Soc. Nephrol. 2017, 28, 2158–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.C.; Moldoveanu, Z.; Julian, B.A.; Novak, J.; Sanders, J.T.; McGlothan, K.R.; Gharavi, A.G.; Wyatt, R.J. Galactose-deficient IgA1 in African Americans with IgA nephropathy: serum levels and heritability. Clin. J. Am. Soc. Nephrol. 2010, 5, 2069–2074. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Ding, J.; Zhu, L.; Shi, S.; Jiang, L.; Zhao, M.; Zhang, H. Aberrant galactosylation of IgA1 is involved in the genetic susceptibility of Chinese patients with IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3372–3375. [Google Scholar] [CrossRef] [Green Version]
- Kiryluk, K.; Moldoveanu, Z.; Sanders, J.T.; Eison, T.M.; Suzuki, H.; Julian, B.A.; Novak, J.; Gharavi, A.G.; Wyatt, R.J. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney Int. 2011, 80, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Lomax-Browne, H.J.; Visconti, A.; Pusey, C.D.; Cook, H.T.; Spector, T.D.; Pickering, M.C.; Falchi, M. IgA1 Glycosylation Is Heritable in Healthy Twins. J. Am. Soc. Nephrol. 2017, 28, 64–68. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Z.; Zhang, H.; Liu, X. Aberrant IgA1 Glycosylation in IgA Nephropathy: A Systematic Review. PLoS ONE 2016, 11, e0166700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feehally, J.; Farrall, M.; Boland, A.; Gale, D.P.; Gut, I.; Heath, S.; Kumar, A.; Peden, J.F.; Maxwell, P.H.; Morris, D.L.; et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J. Am Soc. Nephrol. 2010, 21, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; Julian, B.A.; Wyatt, R.J.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet 2011, 43, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Li, M.; Zhang, H.; Low, H.Q.; Wei, X.; Wang, J.Q.; Sun, L.D.; Sim, K.S.; Li, Y.; Foo, J.N.; et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 44, 178–182. [Google Scholar] [CrossRef]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef]
- Li, M.; Foo, J.N.; Wang, J.Q.; Low, H.Q.; Tang, X.Q.; Toh, K.Y.; Yin, P.R.; Khor, C.C.; Goh, Y.F.; Irwan, I.D.; et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat. Commun. 2015, 6, 7270. [Google Scholar] [CrossRef]
- Kiryluk, K.; Li, Y.; Moldoveanu, Z.; Suzuki, H.; Reily, C.; Hou, P.; Xie, J.; Mladkova, N.; Prakash, S.; Fischman, C.; et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet 2017, 13, e1006609. [Google Scholar] [CrossRef] [Green Version]
- Saka, S.; Hirawa, N.; Oka, A.; Yatsu, K.; Hirukawa, T.; Yamamoto, R.; Matsusaka, T.; Imai, E.; Narita, I.; Endoh, M.; et al. Genome-wide association study of IgA nephropathy using 23 465 microsatellite markers in a Japanese population. J. Hum. Genet. 2015, 60, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Jeong, K.H.; Kim, J.S.; Lee, Y.H.; Kim, Y.G.; Moon, J.Y.; Kim, S.K.; Kang, S.W.; Kim, T.H.; Lee, S.H.; Kim, Y.H.; et al. Genome-wide association study identifies new susceptible loci of IgA nephropathy in Koreans. BMC Med. Genomics 2019, 12, 122. [Google Scholar] [CrossRef]
- Raska, M.; Moldoveanu, Z.; Suzuki, H.; Brown, R.; Kulhavy, R.; Andrasi, J.; Hall, S.; Vu, H.L.; Carlsson, F.; Lindahl, G.; et al. Identification and characterization of CMP-NeuAc:GalNAc-IgA1 alpha2,6-sialyltransferase in IgA1-producing cells. J. Mol. Biol. 2007, 369, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Raska, M.; Yamada, K.; Moldoveanu, Z.; Julian, B.A.; Wyatt, R.J.; Tomino, Y.; Gharavi, A.G.; Novak, J. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J. Biol. Chem. 2014, 289, 5330–5339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, S.M.; Papista, C.; Chemouny, J.M.; Berthelot, L.; Monteiro, R.C. Role of IgA receptors in the pathogenesis of IgA nephropathy. J. Nephrol. 2016, 29, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, A.; van Egmond, M. IgA and FcαRI: Pathological Roles and Therapeutic Opportunities. Front. Immunol. 2019, 10, 553. [Google Scholar] [CrossRef] [PubMed]
- Grossetête, B.; Launay, P.; Lehuen, A.; Jungers, P.; Bach, J.F.; Monteiro, R.C. Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: evidence for a negative regulatory role of serum IgA. Kidney Int. 1998, 53, 1321–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Magistroni, R.; D’Agati, V.D.; Appel, G.B.; Kiryluk, K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015, 88, 974–989. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, R.M.; Radl, J.; Haaijman, J.J.; Vermeer, B.J.; Weening, J.J.; Kauffmann, R.H.; Daha, M.R.; van Es, L.A. Circulating and mesangial secretory component-binding IgA-1 in primary IgA nephropathy. Kidney Int. 1984, 26, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Oortwijn, B.D.; Rastaldi, M.P.; Roos, A.; Mattinzoli, D.; Daha, M.R.; van Kooten, C. Demonstration of secretory IgA in kidneys of patients with IgA nephropathy. Nephrol. Dial. Transplant. 2007, 22, 3191–3195. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Chen, X.; Nishi, S.; Narita, I.; Gejyo, F. Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy. Kidney Int. 2004, 65, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Oortwijn, B.D.; Eijgenraam, J.W.; Rastaldi, M.P.; Roos, A.; Daha, M.R.; van Kooten, C. The role of secretory IgA and complement in IgA nephropathy. Semin. Nephrol. 2008, 28, 58–65. [Google Scholar] [CrossRef]
- Rizk, D.V.; Maillard, N.; Julian, B.A.; Knoppova, B.; Green, T.J.; Novak, J.; Wyatt, R.J. The Emerging Role of Complement Proteins as a Target for Therapy of IgA Nephropathy. Front. Immunol. 2019, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. Biomarkers for IgA nephropathy on the basis of multi-hit pathogenesis. Clin. Exp. Nephrol. 2019, 23, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, M.C.; Moldoveanu, Z.; Suzuki, H.; Berthoux, F.; Julian, B.A.; Sanders, J.T.; Renfrow, M.B.; Novak, J.; Wyatt, R.J. Biomarkers in IgA nephropathy: relationship to pathogenetic hits. Expert Opin. Med. Diagn. 2013, 7, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaskandan, H.; Pawluczyk, I.; Barratt, J. MicroRNAs: A new avenue to understand, investigate and treat immunoglobulin A nephropathy? Clin. Kidney J. 2018, 11, 29–37. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| SERUM IgA | MUCOSAL IgA | |
|---|---|---|
| Production location | Plasmablast and plasma cells in the bone marrow, spleen, lymph nodes, tonsils [12] | Plasmablast and plasma cells located in the lamina propria of mucosal system (MALT, PP, ILF etc.) [20] |
| Production | ≈ 1.2 g per day (adult human) | ≈ 3.2 g per day |
| Form | 75–90% monomeric, [12] 10–15% in polymeric form, [12] 1% bound in circulating immune complexes | Mucosa: 95% SIgA [12] lamina propria: predominantly pIgA, scarce mIgA |
| Subclass | 85% IgA1, 12% IgA2 [12] 101 ± 26 (IgA1) and 14 ± 4 (IgA2) mg/kg body weight | % of IgA1 and IgA2 varies and depends on the mucosal area, i.e., tears, saliva, respiratory mucosa, vaginal, genital intestine (see Figure 1) |
| Function | Anti-inflammatory effects: binding of mIgA to FcαRI induce inhibitory effects and downregulates IgG-mediated phagocytosis, chemotaxis, bactericidal activity, oxidative burst activity, and cytokine release [21]. | Bacteriostatic activity Barrier for microbiota (pathogens, and commensal bacteria), toxins from crossing the epithelial layer; neutralization of intracellular pathogens [13,18] |
| Clearance | Catabolized in liver, kidney, skin; a half-life ~5 days. Phagocytosis of IgA-Ag complex [22] | Secreted into lumen (excreted) Phagocytosis of IgA-Ag complex when in lamina propria or intraepithelial |
| Receptor | Recognition Site (Ig Type, Form) | Cell Type Expressing the Receptor | Function | Ref |
|---|---|---|---|---|
| pIgR | J-chain (pIgA, pIgM) | Secretory epithelial cells in the intestine, salivary glands, bronchial mucosa, mammary glands, uterine | Transport of pIgA from lamina propria across the epithelial cells to secretions lumen, where it is released as SIgA (part of the receptor becomes part of SIgA; secretory component) | [30] |
| Epithelial cells of biliary duct | Transport of serum pIgA into the bile (which excretes into intestinal lumen) | |||
| DC-SIGN | N-/O-linked glycans (SIgA) | Dendritic cell Cell culture | Binding and internalizing SIgA | [31] |
| Dectin-1 Siglec-5 | Cα1 and Cα2 (SIgA) | M cell | IgA-specific receptor on the apical surface that mediates the transepithelial transport to GALT | [28,32] |
| ASGPR | O-linked glycans on hinge region - terminal Gal or GalNAc of desialylated O-glycoproteins (free IgA1) | Hepatocytes | Clearance of IgA1 from the circulation and catabolic degradation (lysosomal catabolism) | [12,23,33,34] |
| Monocytes | A mobile pool of the receptors, capable of reaching sites remote from the liver | |||
| Soluble form in circulation | Bind to free IgA1 in the circulation and transport to liver for uptake and degradation by hepatocytes [34] | |||
| TfR TfR1 (CD71) | (IgA1; Monomeric better than polymeric) | Mesangial cells CD71 is expressed on a wide range of tissues TfR2 predominantly in the liver | Clearance of IgA from the circulation Binding of IgA1 to TfR1 depends on the size and glycosylation of IgA1 and can be inhibited by transferring | [24] |
| β-GalT1 | Fc region | Mesangial cells | Clearance of IgA from the circulation | [25] |
| Leukocyte receptors | ||||
| SCR | Secretory component (SIgA, SC) | Eosinophils basophils | Generate respiratory burst and eosinophil degranulation, target killing, and release of proinflammatory cytokines and other mediators | [27,35] |
| FcRL4 | Fc region (IgA) | Memory B cells in mucosal lymphoid tissue | Immune complex-dependent B cell regulation | [29] |
| Fcα/μR (CD351) | Fc region (IgA, IgM; Polymeric and immune complexes) | Mature B cells, macrophages Constitutively express | Endocytosis of IgA/IgM-coated microbes, phagocytosis higher affinity for IgM than IgA (10x) | [26] |
| FcαRI (CD89) | Fc region; CHα2 and CHα3, (immune complexes and pIgA better than monomeric SIgA only with lectin Mac-1) | Neutrophils, eosinophils, monocytes, Kupffer cells, macrophages, subpopulation of T and B cells, subset of DC, NK | Bifunctional receptor – the function depends on IgA ligand avidity: Anti-inflammatory: (free mIgA) inhibition of phagocytic activity, stimulate release of anti-inflammatory cytokines by myeloid cells Proinflammatory: (IgA + Ag complex receptor cross-linking): stimulation of phagocytosis, respiratory burst, release of ROS and proinflammatory cytokines, antigen presentation, antibody-dependent cellular cytotoxicity | [36,37] |
| Soluble form of CD89 in circulation | Binds CD71 and induces TGase 2, which in turn is translocated to the mesangial plasma membrane allowing cell activation by IgA1-sCD89 complexes | |||
| Sample Size (Population) | Mesangial Deposition | Positive Cases | Clinical or Histological Features of Cases with IgA Deposits | Ref |
|---|---|---|---|---|
| Primary glomerular IgA deposition in individuals without clinical manifestation of renal disease | ||||
| 510 kidney transplant cases (64 cadaveric and 446 living donors) (Japanese donors) | IgA IgA + C3 | 82/510 (16%) 16/82 | IgA + C3 deposition was associated with mild degree of microhematuria, mesangial proliferation, and glomerular macrophage infiltration | [38] |
| 756 autopsy cases (violent death) (Finland) | IgA IgA + C3b IgA + C1q IgA + IgG | 52/756 (6.8%) 4/52 2/52 8/52 | 10/52 cases had morphological changes suggestive of renal disease | [39] |
| 200 autopsy cases (violent death) (Singapore) | IgA IgA + C3b IgA + C1q IgA + IgG | 8/200 (4%) 2/8 0/8 4/8 | Histology revealed only minimal morphological alterations | [40] |
| Secondary glomerular IgA deposition | ||||
| 250 consecutive autopsy cases (non-selected) (Germany) | IgA IgA + C3 IgA + C5 IgA + IgG(+) | 12/250 (4.8%) 1/6 5/6 2/6 | 6/12 associated with liver cirrhosis 6/12 associated with endocarditis, bronchial asthma, cardiovascular disease, or neoplasia; IgA1+, IgA2 (+), SC (+) | [41] |
| Sample Population | Controls Healthy Unrelated | Ancestry of Patients | Heritability (P Value) | Ref |
|---|---|---|---|---|
| 89 adult IgAN patients vs. 266 blood relatives | 150 adults | European | 0.54 (0.0001) | [46] |
| 63 adult IgAN patients vs. 32 first-degree relatives | 44 adults | Chinese Asian | Yes nd | [48] |
| 11 pediatric and 18 adult IgAN patients vs. 34 first-degree relatives | 45 pediatric (European) 150 adult (European) | African American | 0.74 (0.007) | [47] |
| 14 pediatric IgAN patients vs. 25 first-degree relatives | 51 pediatric 141 adults | European | 0.76 (<0.05) | [49] |
| 134 adult IgAN trios | 638 adults | UK whites | 0.387 (<0.05) | [45] |
| 20 pediatric HSPN patients vs. 28 first-degree relatives | 51 pediatric 141 adults | European | 0.64 (<0.05) | [49] |
| 27 monozygotic healthy female twin pairs 47 dizygotic healthy female twin pairs | European (UK) | 0.84 0.46 (<0.05) | [50] | |
| GWAS | Sample Population | Ancestry of Patients | Identified Loci | IgAN Risk |
|---|---|---|---|---|
| [52] | 914 cases vs. 5069 controls | European | 6p–HLA-B, DRB1, DQA, DQB | nr |
| [53] | 3144 cases vs. 2822 controls | Chinese, European | 1q32-CFHR1, CFHR3; 22q12.2–OSM, LIF, HORMAD2, MTMR3; 6p21–HLA-DRB1, HLA-DQB1, HLA-DPA1, HLA-DPB1, HLA-DPB2, TAP2, TAP1, PSMB8, PSMB9 | 4–7% |
| [54] | 4137 cases vs. 7734 controls | Chinese | 8p23–DEFA, 17p13-TNFSF13; 22q12–HORMAD2 | nr |
| [55] | 7658 cases vs. 12,954 controls | European, East Asian | 1p13-VAV3; 9q34–CARD9; 16p11-ITGAM, ITGAX; 8p23–DEFA; 6p21–HLA-DQ-HLA-DR, TAP1–PSMB8 and HLA-DP; 1q32-CFHR1, CFHR3; 17p13-TNFSF13; 22q12–HORMAD2 | 5% |
| [56] | 8313 cases vs. 19,680 controls | Chinese | 3q27.3-ST6GAL1; 8p23–DEFA; 11p11.2-ACCS; 8q22.3-ODF1-KLF10; 16p11-ITGAM, ITGAX | 1.7% 5% |
| [57] | 2633 cases | European, East Asian | Xq24-C1GALT1C1; 7p21.3-C1GALT1 | 7% 2% |
| [58] | 915 patients vs. 481 controls | Japanese | 6p21–HLA locus; 12q12–TSPAN8-PTPRR locus | nr |
| [59] | 498 patients vs. 893 controls | Koreans | 10p15.1-ANKRD16 | nr |
| Locus | Gene | Function |
|---|---|---|
| 1p13 | VAV3 | Chemokine signaling; NK, T cells, B cells, FcεRI, FcγR. VAV proteins are essential for adaptive immune function and NF-κB activation in B cells, i.e., a process that stimulates IgA production |
| 1q32 | CFHR1, CFHR3 | Complement system; encode Factor H related peptides that modulate the activity of the alternative complement pathway. FHR1 competes with factor H for binding to surface-fixed C3b leading to activation of C3 convertase |
| 6p21 | HLA-DQA1, HLA-DQB1, HLA-DRB1 PSMB8, PSMB9, TAP1, TAP2 | MHC class II molecules critical for antigen processing and presentation and adaptive immunity Phagosome pathway |
| 8p23 | DEFA1, DEFA3, DEFA4, DEFA5, DEFA6 | Innate immunity; antimicrobial peptides in mucosal defense; α-defensins 1,3,4 are synthesized in neutrophils, while α-defensins 5 and 6 are constitutively produced by the Paneth cells in the small intestine |
| 8q22.3 | ODF1-KLF10 | Encodes a transcriptional repressor that acts as an effector of TGFβ signaling |
| 9q34 | CARD9 | Innate immunity; NOD-like receptor signaling |
| 11p11.2 | ACCS | Encodes a1-aminocyclopropane-1-carboxylate synthase homologue that interact with a protein required for epithelial cell polarization and ciliogenesis |
| 16p11 | ITGAM, ITGAX | Encode leukocyte-specific α-integrins involved in the process of phagocytosis and regulation of IgA production |
| 17p13 | TNFSF13 | Encode APRIL, a B cell stimulating cytokine induced by intestinal bacteria and promotes CD40-independent IgA class switching |
| 22q12 | LIF, OSM, HORMAD2, MTMR3 | Cytokine-cytokine interaction; cytokine encoding genes expressed in mucosal tissues with immunomodulatory properties JAK-STAT signaling pathway |
| 3q27.3 | ST6GAL1 | Encode enzyme ST3Gal-1 responsible for sialylation of Gal |
| 7p21.3 | C1GALT1 | Encode enzyme C1GalT1 that catalyzes attachment of Gal to GalNAc |
| Xq24 | C1GALT1C1 | Encode chaperone Cosmc, required for the stability of C1GalT1 enzyme |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perše, M.; Večerić-Haler, Ž. The Role of IgA in the Pathogenesis of IgA Nephropathy. Int. J. Mol. Sci. 2019, 20, 6199. https://doi.org/10.3390/ijms20246199
Perše M, Večerić-Haler Ž. The Role of IgA in the Pathogenesis of IgA Nephropathy. International Journal of Molecular Sciences. 2019; 20(24):6199. https://doi.org/10.3390/ijms20246199
Chicago/Turabian StylePerše, Martina, and Željka Večerić-Haler. 2019. "The Role of IgA in the Pathogenesis of IgA Nephropathy" International Journal of Molecular Sciences 20, no. 24: 6199. https://doi.org/10.3390/ijms20246199
APA StylePerše, M., & Večerić-Haler, Ž. (2019). The Role of IgA in the Pathogenesis of IgA Nephropathy. International Journal of Molecular Sciences, 20(24), 6199. https://doi.org/10.3390/ijms20246199