Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction

 ,
,
Abstract
:1. Introduction
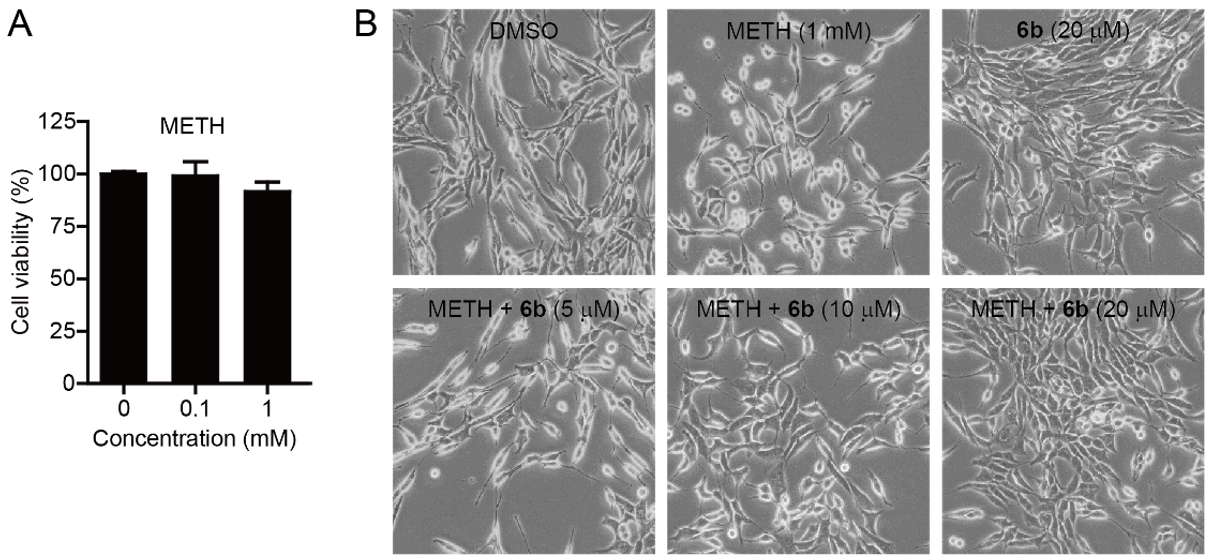
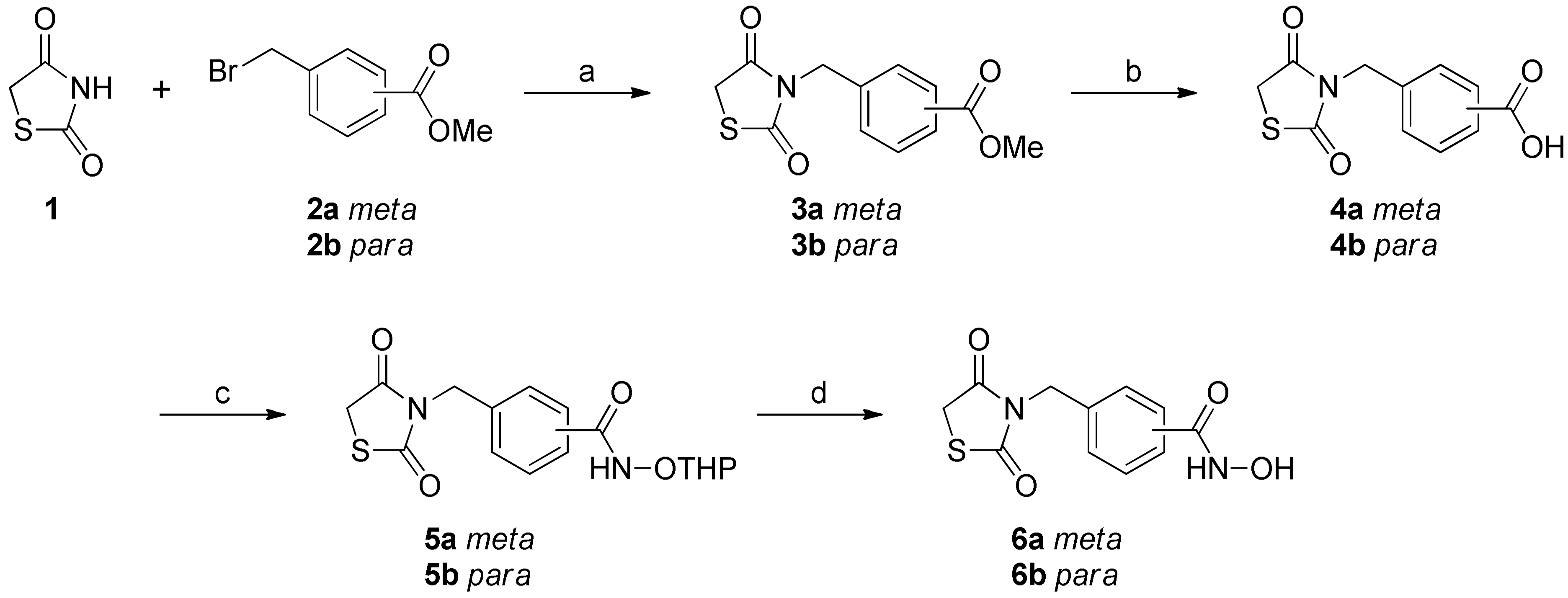
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods and Materials
3.1.2. General Procedure for the Synthesis of Compounds 3a-b
Methyl 3-((2,4-dioxothiazolidin-3-yl)methyl)benzoate (3a)
Methyl 4-((2,4-dioxothiazolidin-3-yl)methyl)benzoate (3b)
3.1.3. General Procedure for the Synthesis of Compounds 4a-b
3-((2,4-Dioxothiazolidin-3-yl)methyl)benzoic acid (4a)
4-((2,4-Dioxothiazolidin-3-yl)methyl)benzoic acid (4b)
3.1.4. General Procedure for the Synthesis of Compounds 5a-b
3-((2,4-Dioxothiazolidin-3-yl)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (5a)
4-((2,4-Dioxothiazolidin-3-yl)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (5b)
3.1.5. General Procedure for the Synthesis of Compounds 7a-c
4-((2,4-Dioxo-5-propylthiazolidin-3-yl)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (7a)
4-((5-Allyl-2,4-dioxothiazolidin-3-yl)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (7b)
4-((5-Benzyl-2,4-dioxothiazolidin-3-yl)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (7c)
3.1.6. General Procedure for the Synthesis of Compounds 6a-b and 8a-c
3-((2,4-Dioxothiazolidin-3-yl)methyl)-N-hydroxybenzamide (6a)
4-((2,4-Dioxothiazolidin-3-yl)methyl)-N-hydroxybenzamide (6b)
4-((2,4-Dioxo-5-propylthiazolidin-3-yl)methyl)-N-hydroxybenzamide (8a)
4-((5-Allyl-2,4-dioxothiazolidin-3-yl)methyl)-N-hydroxybenzamide (8b)
4-((5-Benzyl-2,4-dioxothiazolidin-3-yl)methyl)-N-hydroxybenzamide (8c)
3.2. Biology
3.2.1. Materials
3.2.2. Cell Culture
3.2.3. Assessment of Cell Morphology
3.2.4. HDAC Assay
3.2.5. Cell Proliferation Assay
3.2.6. Western Blot
3.2.7. Statistical Analysis
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Galbraith, N. The methamphetamine problem: Commentary on psychiatric morbidity and socio-occupational dysfunction in residents of a drug rehabilitation centre. BJPsych Bull. 2015, 39, 218–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, K.E.; Ray, L.A. Methamphetamine: An update on epidemiology, pharmacology, clinical phenomenology, and treatment literature. Drug Alcohol Depend. 2014, 143, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, H.; Killinger, B.A.; Miller, C.V.; Moszczynska, A. Single and binge methamphetamine administrations have different effects on the levels of dopamine D2 autoreceptor and dopamine transporter in rat striatum. Int. J. Mol. Sci. 2014, 15, 5884–5906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Chen, Y.Y.; Shen, Y.; Cao, X.; Li, A.; Liu, Q.; Li, Z.; Zhang, L.B.; Dai, W.; Tan, T.; et al. Methamphetamine abuse impairs motor cortical plasticity and function. Mol. Psychiatry 2017, 22, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.L.; Bian, J.W.; Zheng, Z.J.; Zhao, L.; Han, S.; Sun, X.H.; Li, J.F.; Ni, G.X. Effects of methamphetamine abuse on spatial cognitive function. Sci. Rep. 2018, 8, 5502. [Google Scholar] [CrossRef] [PubMed]
- Groman, S.M.; Rich, K.M.; Smith, N.J.; Lee, D.; Taylor, J.R. Chronic Exposure to Methamphetamine Disrupts Reinforcement-Based Decision Making in Rats. Neuropsychopharmacology 2018, 43, 770–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Lee, J.H.; Seo, Y.H.; Jang, J.H.; Jeong, C.H.; Lee, S.; Jeong, G.S.; Park, B. Epicatechin Prevents Methamphetamine-Induced Neuronal Cell Death via Inhibition of ER Stress. Biomol. Ther. (Seoul) 2019, 27, 145–151. [Google Scholar] [CrossRef]
- Krasnova, I.N.; Cadet, J.L. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009, 60, 379–407. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.M.; Penrod, R.D.; Barry, S.M.; Hughes, B.W.; Taniguchi, M.; Cowan, C.W. It is a complex issue: Emerging connections between epigenetic regulators in drug addiction. Eur. J. Neuro. Sci. 2019, 50, 2477–2491. [Google Scholar] [CrossRef]
- Godino, A.; Jayanthi, S.; Cadet, J.L. Epigenetic landscape of amphetamine and methamphetamine addiction in rodents. Epigenetics 2015, 10, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Torres, O.V.; Ladenheim, B.; Jayanthi, S.; McCoy, M.T.; Krasnova, I.N.; Vautier, F.A.; Cadet, J.L. An Acute Methamphetamine Injection Downregulates the Expression of Several Histone Deacetylases (HDACs) in the Mouse Nucleus Accumbens: Potential Regulatory Role of HDAC2 Expression. Neurotox Res. 2016, 30, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L. Epigenetics of Stress, Addiction, and Resilience: Therapeutic Implications. Mol. Neurobiol. 2016, 53, 545–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalda, A.; Heidmets, L.T.; Shen, H.Y.; Zharkovsky, A.; Chen, J.F. Histone deacetylase inhibitors modulates the induction and expression of amphetamine-induced behavioral sensitization partially through an associated learning of the environment in mice. Behav. Brain Res. 2007, 181, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, S.; Salta, S.; Summavielle, T. Methamphetamine promotes alpha-tubulin deacetylation in endothelial cells: The protective role of acetyl-l-carnitine. Toxicol. Lett. 2015, 234, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys Acta. 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Choi, M.A.; Park, S.Y.; Chae, H.Y.; Song, Y.; Sharma, C.; Seo, Y.H. Design, synthesis and biological evaluation of a series of CNS penetrant HDAC inhibitors structurally derived from amyloid-β probes. Sci. Rep. 2019, 9, 13187. [Google Scholar] [CrossRef]
- Lim, J.; Song, Y.; Jang, J.-H.; Jeong, C.-H.; Lee, S.; Park, B.; Seo, Y.H. Aspirin-inspired acetyl-donating HDACs inhibitors. Arch. Pharm. Res. 2018, 41, 967–976. [Google Scholar] [CrossRef]
- Song, Y.; Lim, J.; Seo, Y.H. A novel class of anthraquinone-based HDAC6 inhibitors. Eur. J. Med. Chem. 2019, 164, 263–272. [Google Scholar] [CrossRef]
- Sucheta; Tahlan, S.; Verma, P.K. Biological potential of thiazolidinedione derivatives of synthetic origin. Chem. Cent. J. 2017, 11, 130. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes 1996, 45, 1661–1669. [Google Scholar] [CrossRef]
- Ma, L.; Pei, H.; Lei, L.; He, L.; Chen, J.; Liang, X.; Peng, A.; Ye, H.; Xiang, M.; Chen, L. Structural exploration, synthesis and pharmacological evaluation of novel 5-benzylidenethiazolidine-2,4-dione derivatives as iNOS inhibitors against inflammatory diseases. Eur. J. Med. Chem. 2015, 92, 178–190. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wu, R.; Cui, X.; Zha, L.; Yu, L.; Shi, H.; Xue, B. Histone Deacetylase 1 (HDAC1) Negatively Regulates Thermogenic Program in Brown Adipocytes via Coordinated Regulation of Histone H3 Lysine 27 (H3K27) Deacetylation and Methylation. J. Biol. Chem. 2016, 291, 4523–4536. [Google Scholar] [CrossRef] [Green Version]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [(18)F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | HDAC1 (IC50; nM) | HDAC6 (IC50; nM) | Selectivity Index a |
|---|---|---|---|---|
| 6a |  | >50,000 | 1961 | NA |
| 6b |  | 388 | 21 | 18.5 |
| 8a |  | >100,000 | 79,290 | NA |
| 8b |  | >100,000 | 1302 | NA |
| 8c |  | >100,000 | 628 | NA |
| SAHA |  | 236 | 226 | 1.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, C.; Oh, Y.J.; Park, B.; Lee, S.; Jeong, C.-H.; Lee, S.; Seo, J.H.; Seo, Y.H. Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction. Int. J. Mol. Sci. 2019, 20, 6213. https://doi.org/10.3390/ijms20246213
Sharma C, Oh YJ, Park B, Lee S, Jeong C-H, Lee S, Seo JH, Seo YH. Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction. International Journal of Molecular Sciences. 2019; 20(24):6213. https://doi.org/10.3390/ijms20246213
Chicago/Turabian StyleSharma, Chiranjeev, Yong Jin Oh, Byoungduck Park, Sooyeun Lee, Chul-Ho Jeong, Sangkil Lee, Ji Hae Seo, and Young Ho Seo. 2019. "Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction" International Journal of Molecular Sciences 20, no. 24: 6213. https://doi.org/10.3390/ijms20246213
APA StyleSharma, C., Oh, Y. J., Park, B., Lee, S., Jeong, C. -H., Lee, S., Seo, J. H., & Seo, Y. H. (2019). Development of Thiazolidinedione-Based HDAC6 Inhibitors to Overcome Methamphetamine Addiction. International Journal of Molecular Sciences, 20(24), 6213. https://doi.org/10.3390/ijms20246213