An Integrated Pan-Cancer Analysis and Structure-Based Virtual Screening of GPR15

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
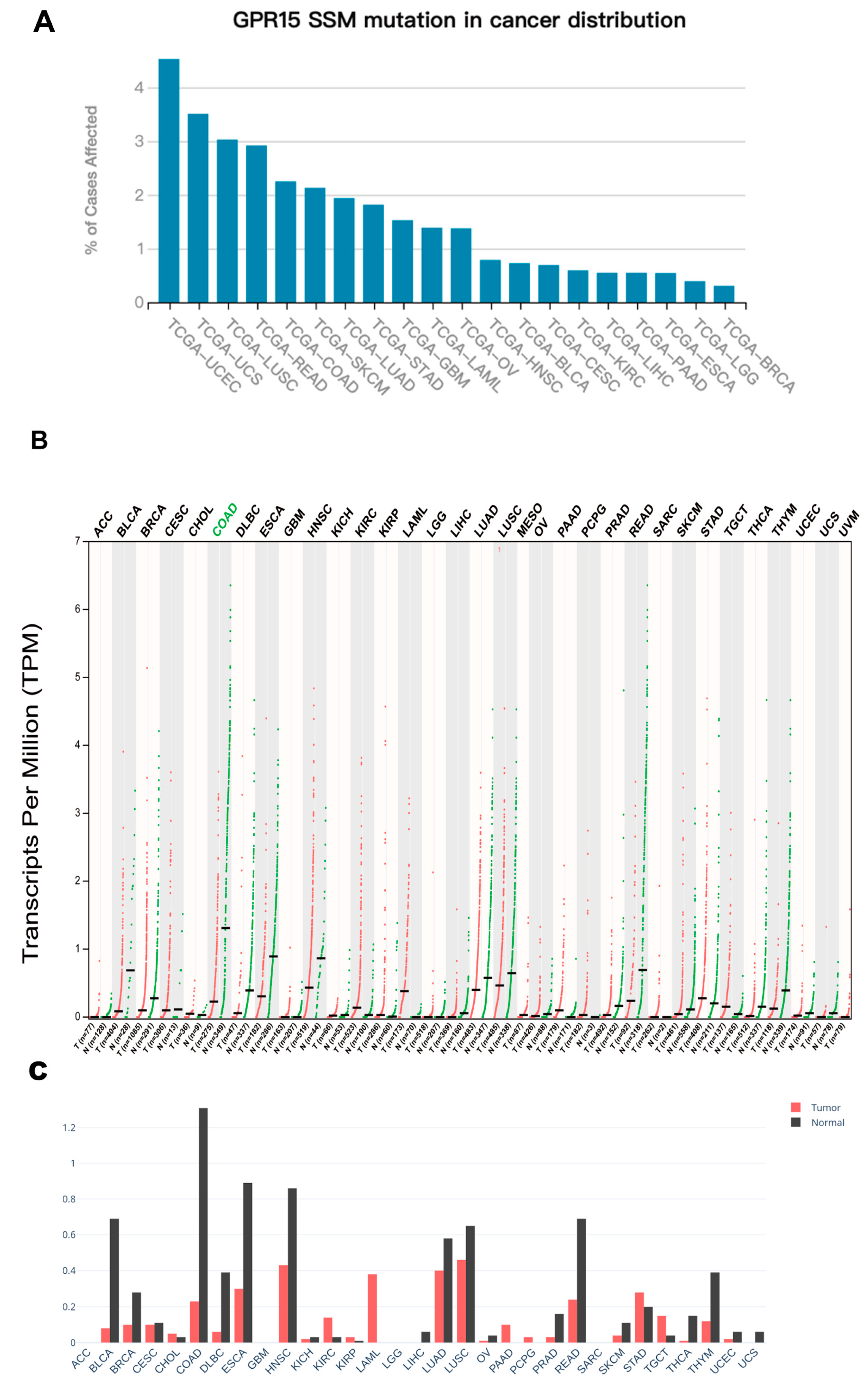
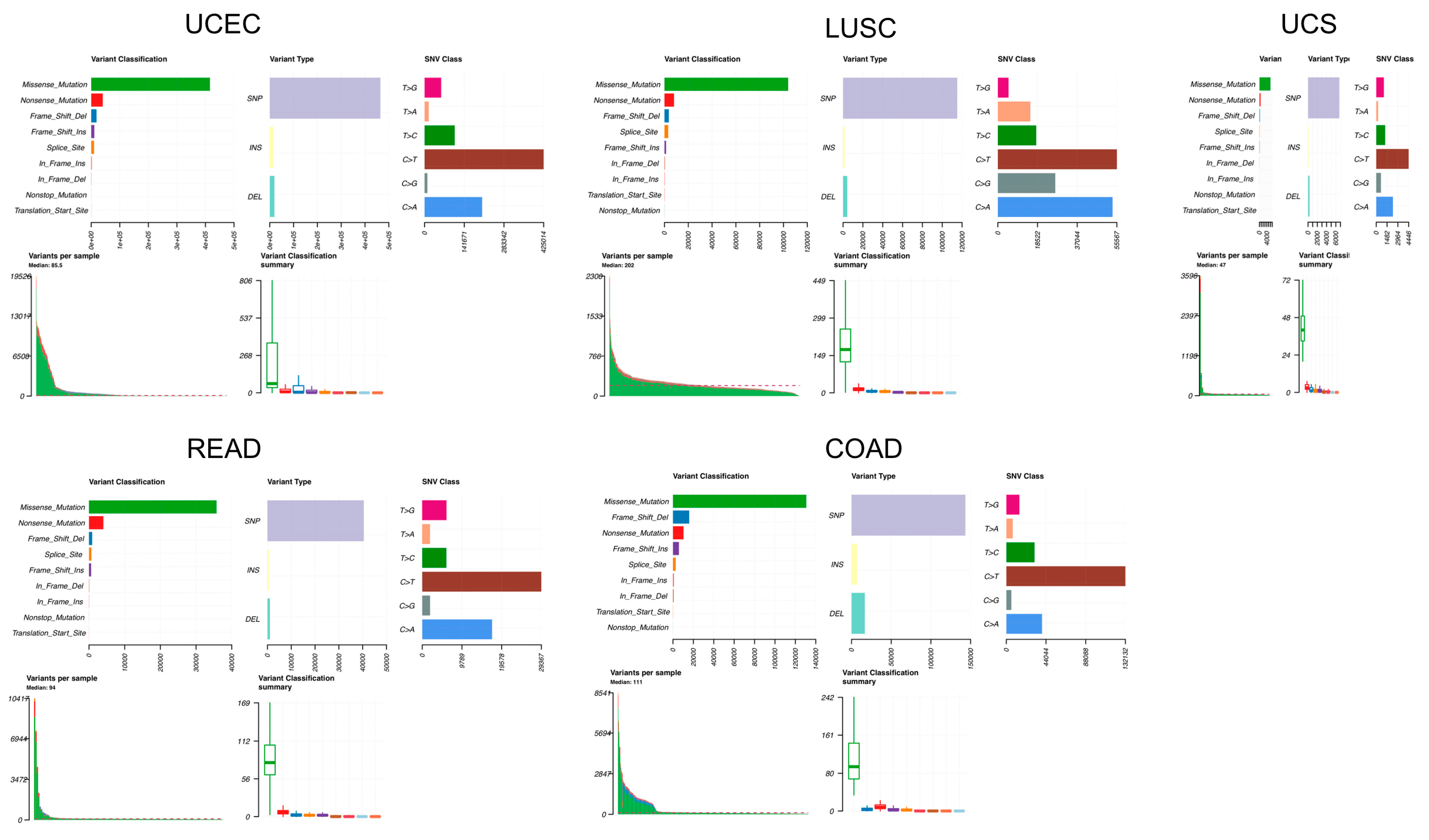
2.1. Pan-Cancer Mutational and Expression Landscape of GPR15
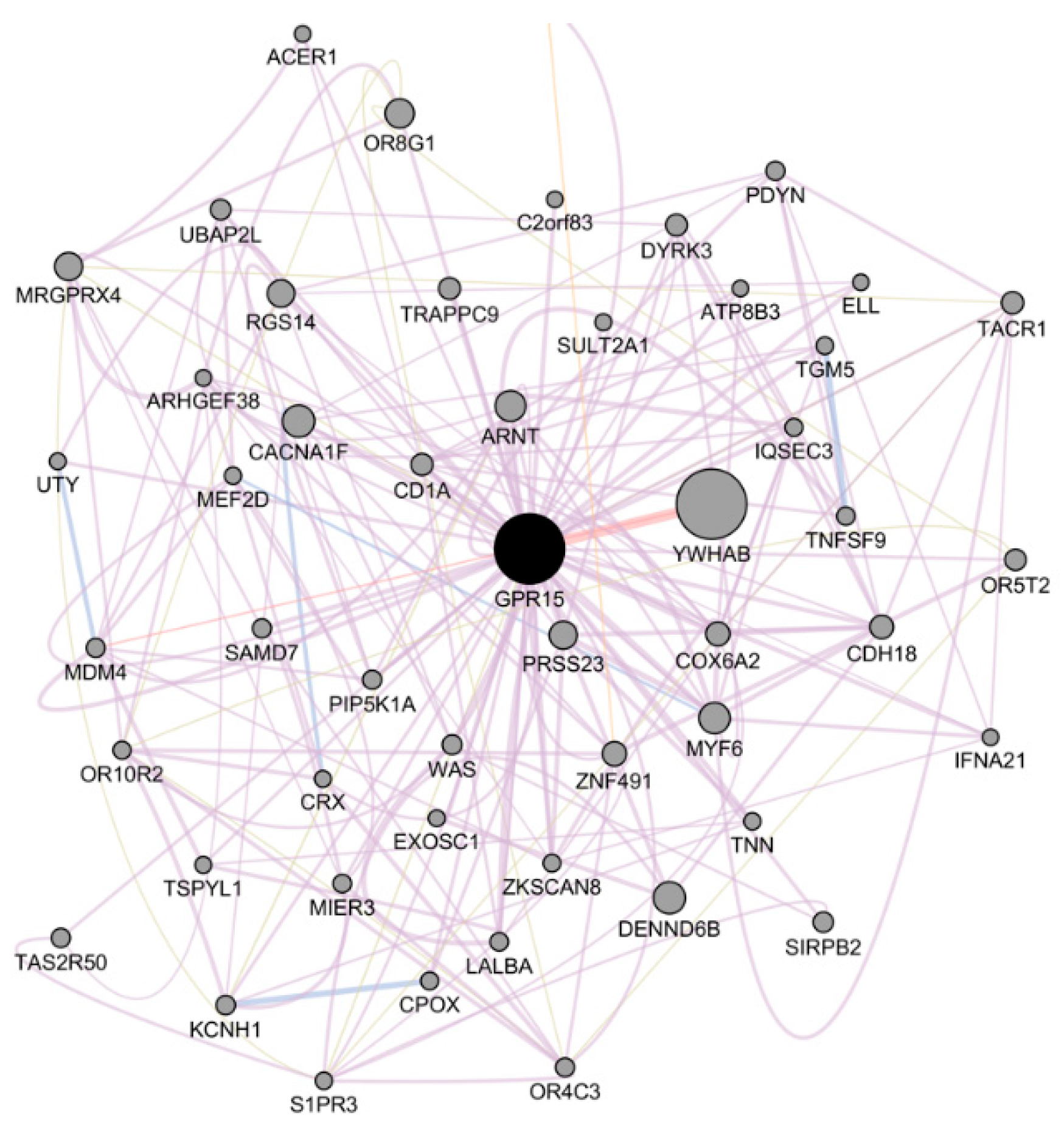
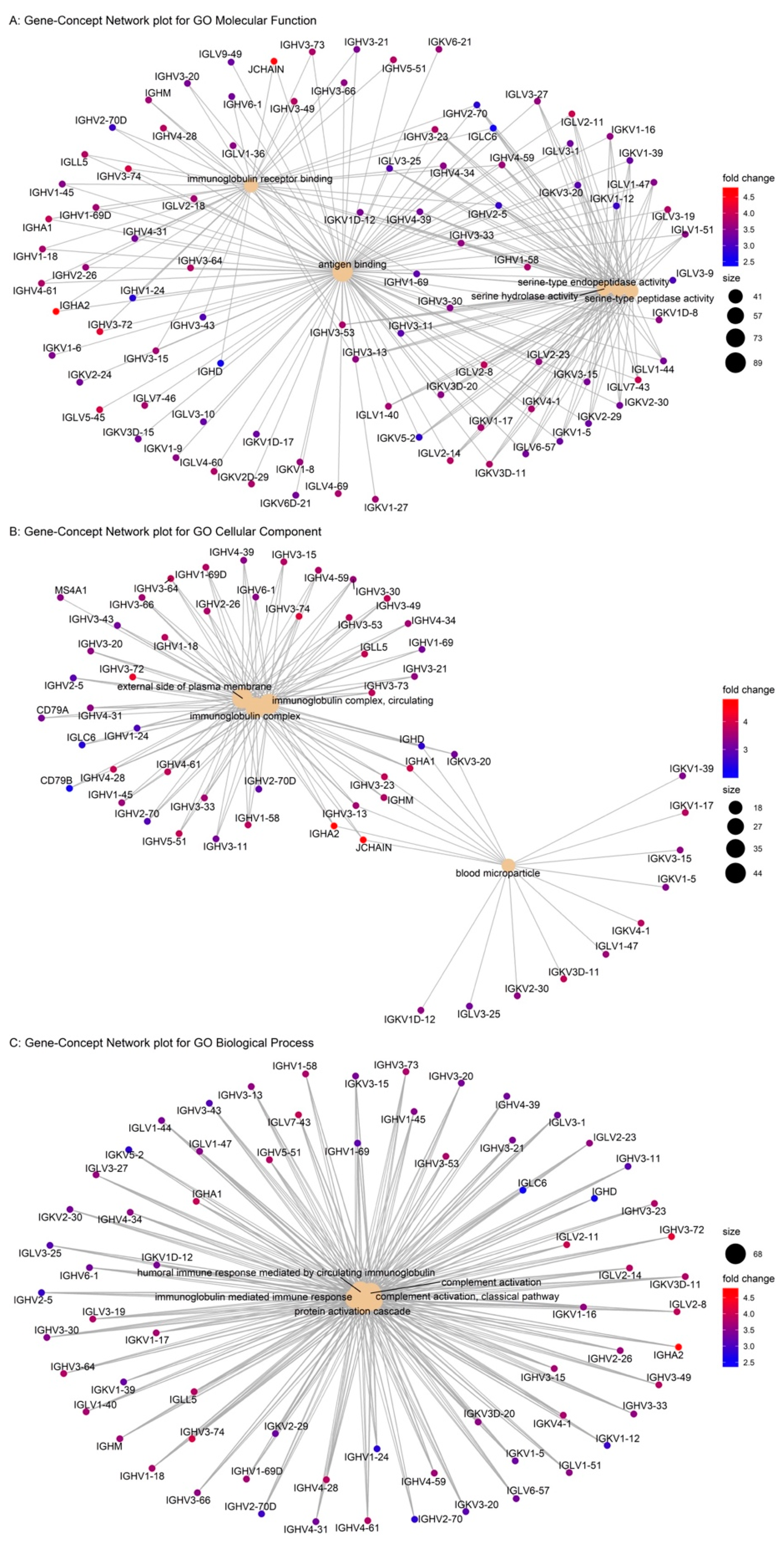
2.2. Integrated Network Analysis of GPR15
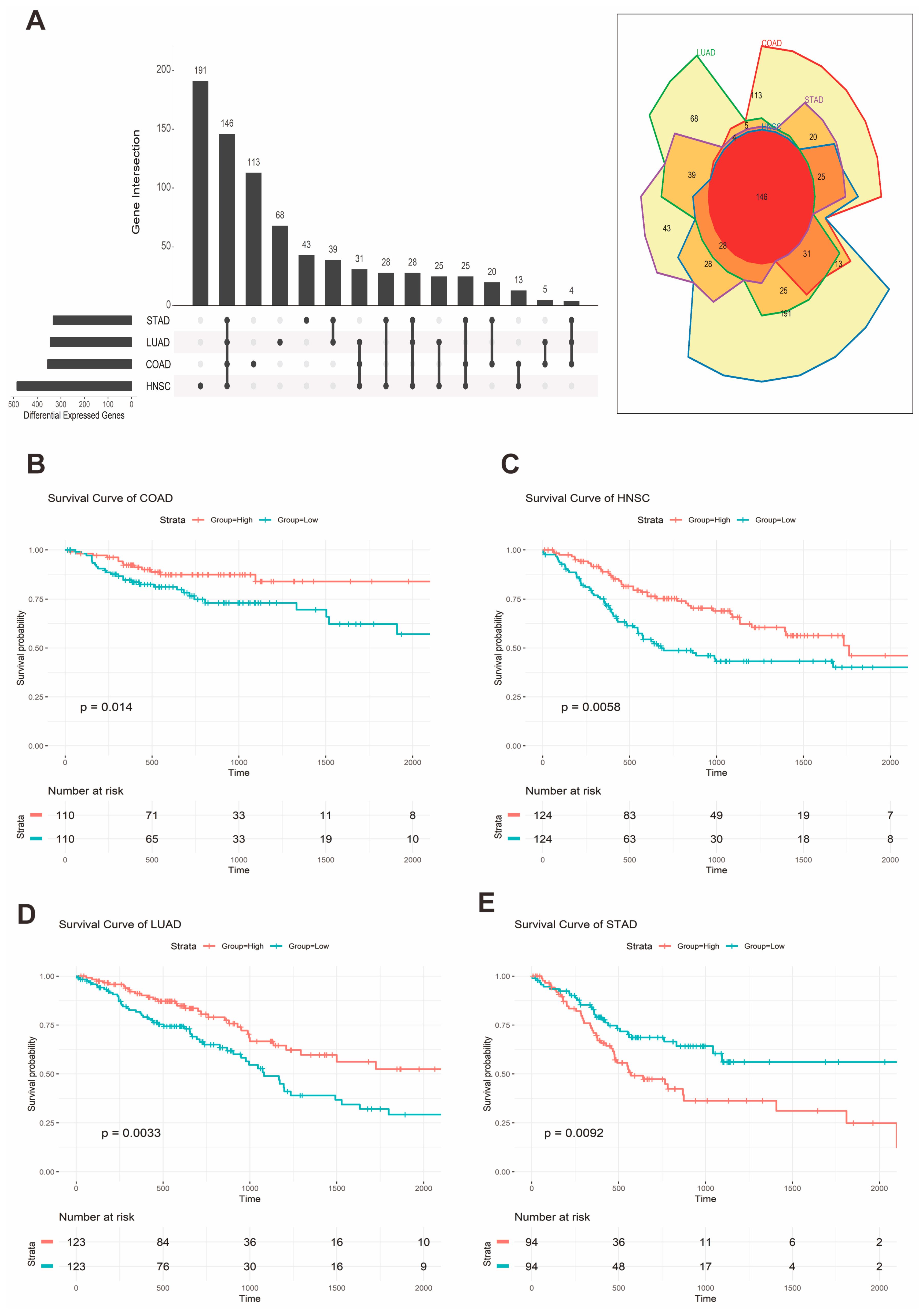
2.3. Pan-Cancer Analysis of GPR15 Expression and Prognostic Association
2.4. Commonly Upregulated Gene Set in High GPR15 Groups of COAD, HNSC, LUAD, and STAD
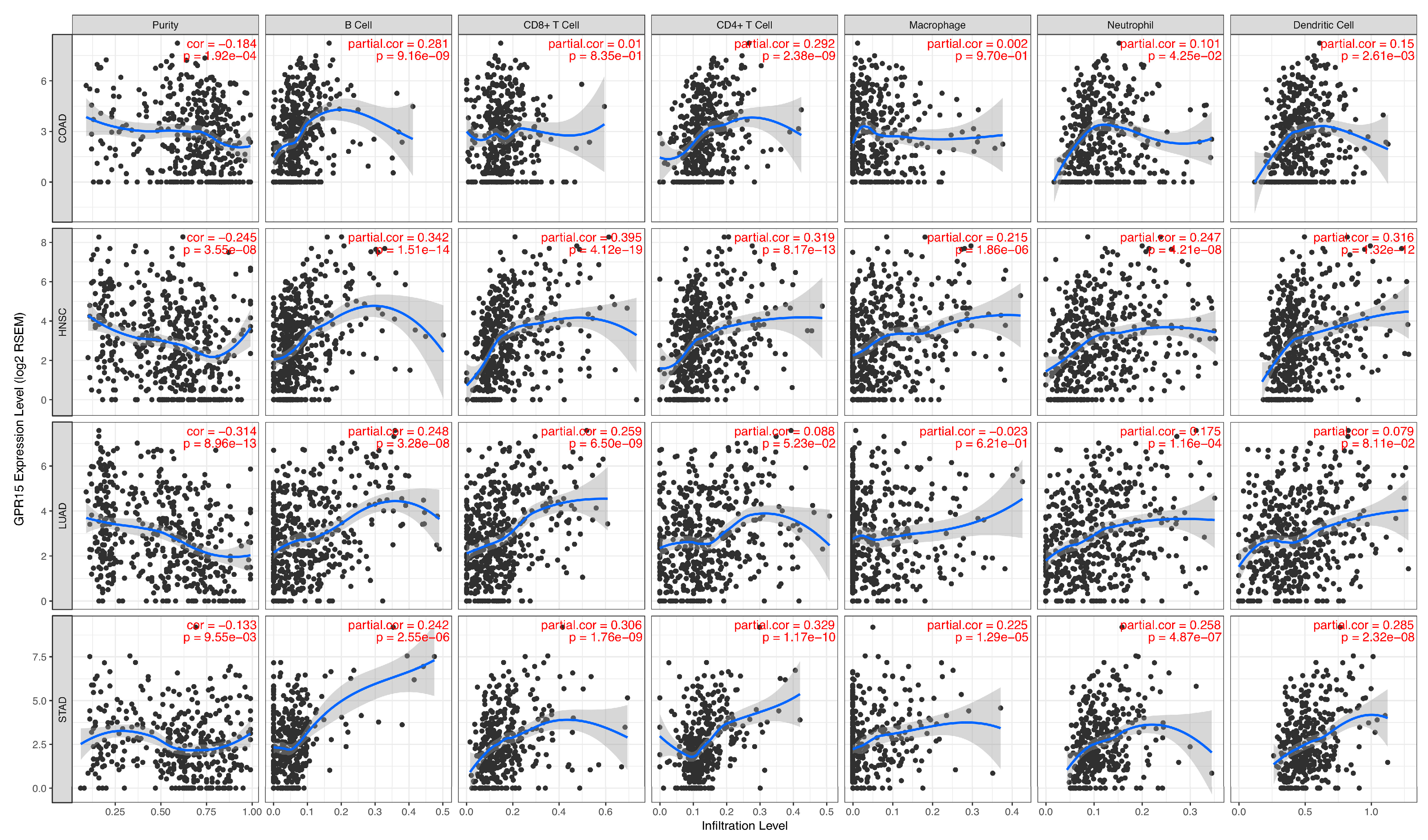
2.5. Association between GPR15 Expression Levels and the Immune Cell-Infiltrating Levels in Cancer
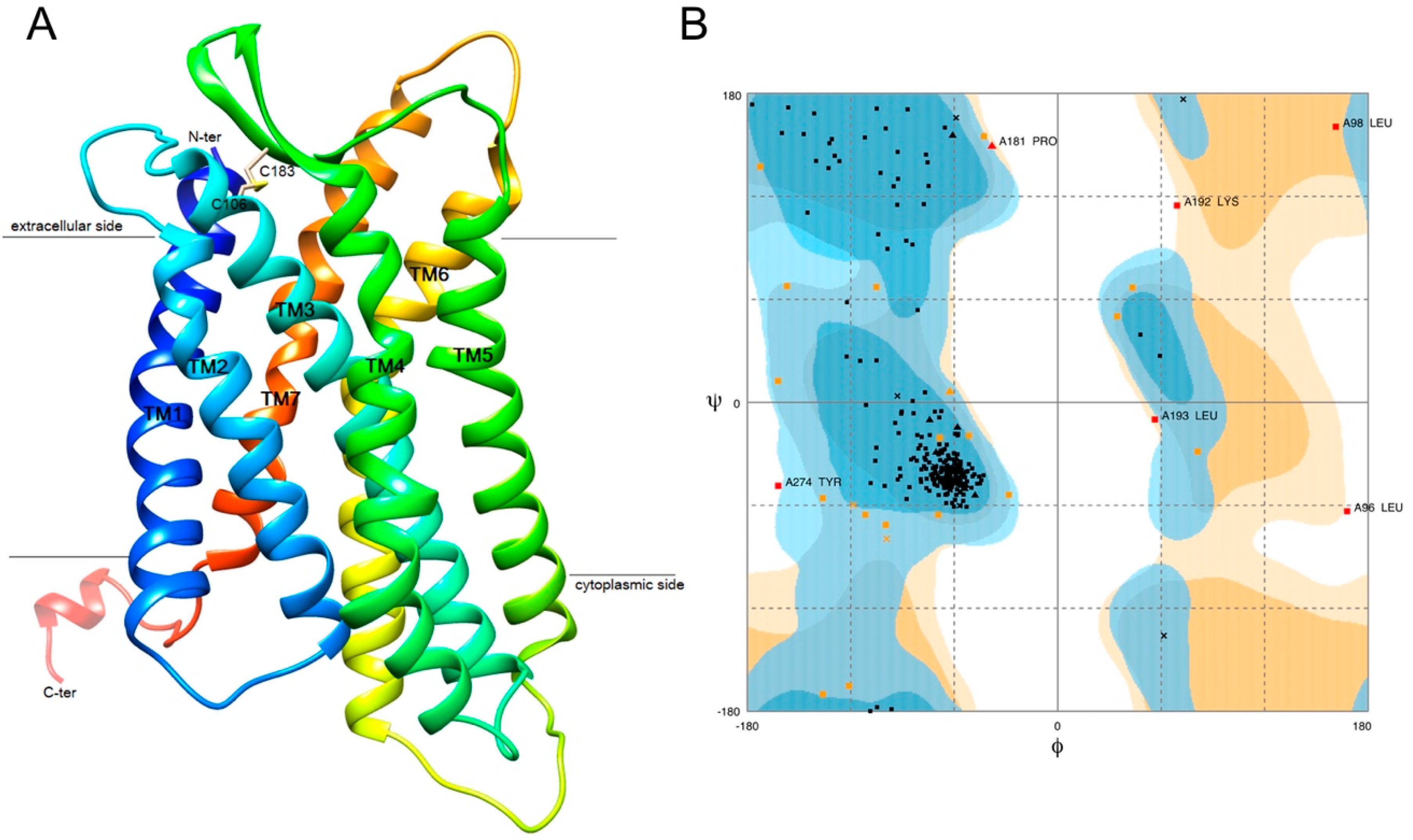
2.6. 3D Structure Modeling of GPR15
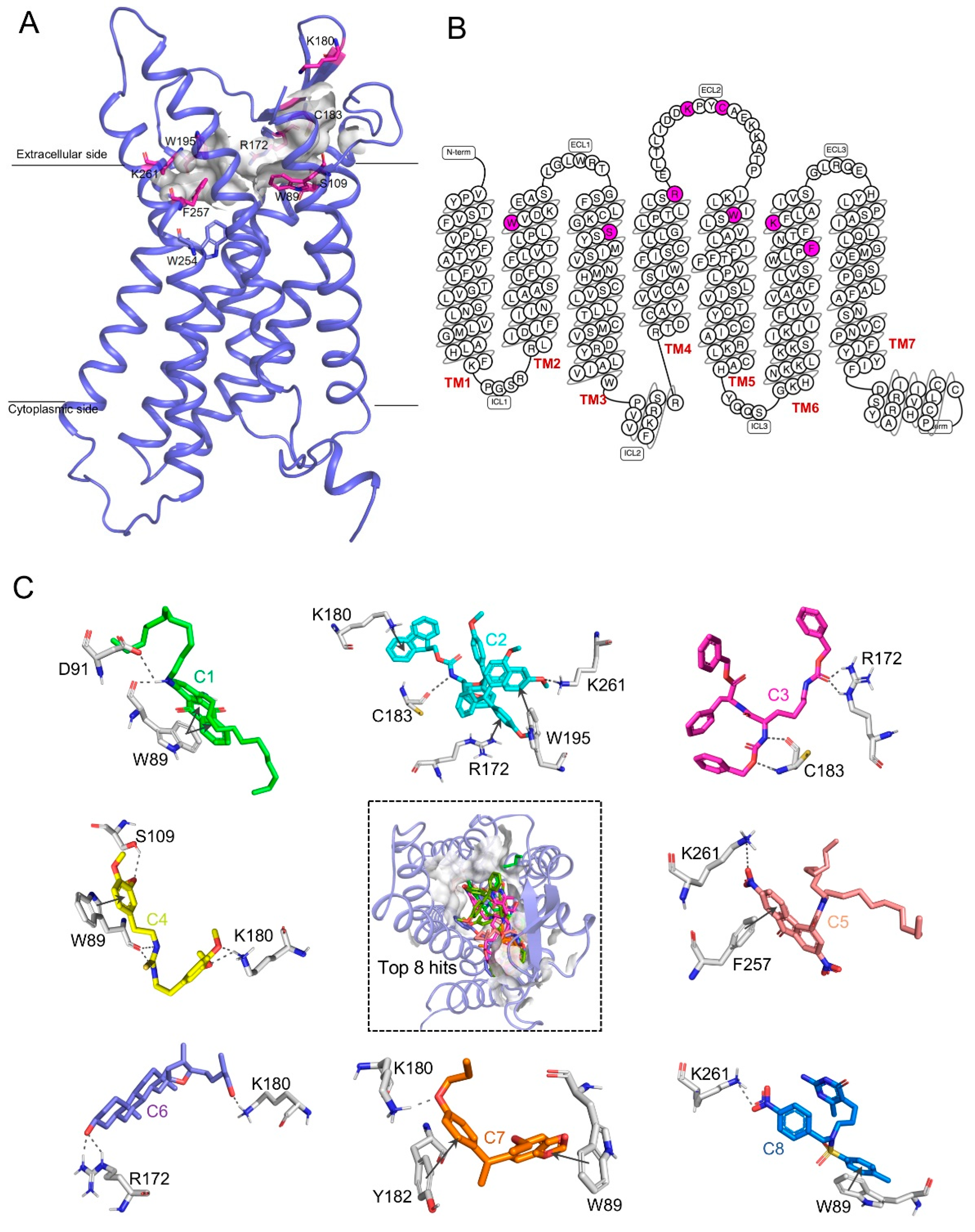
2.7. Structure–Function Relationship-Based Binding Site Prediction
2.8. Virtual Screening and Molecular Docking Results
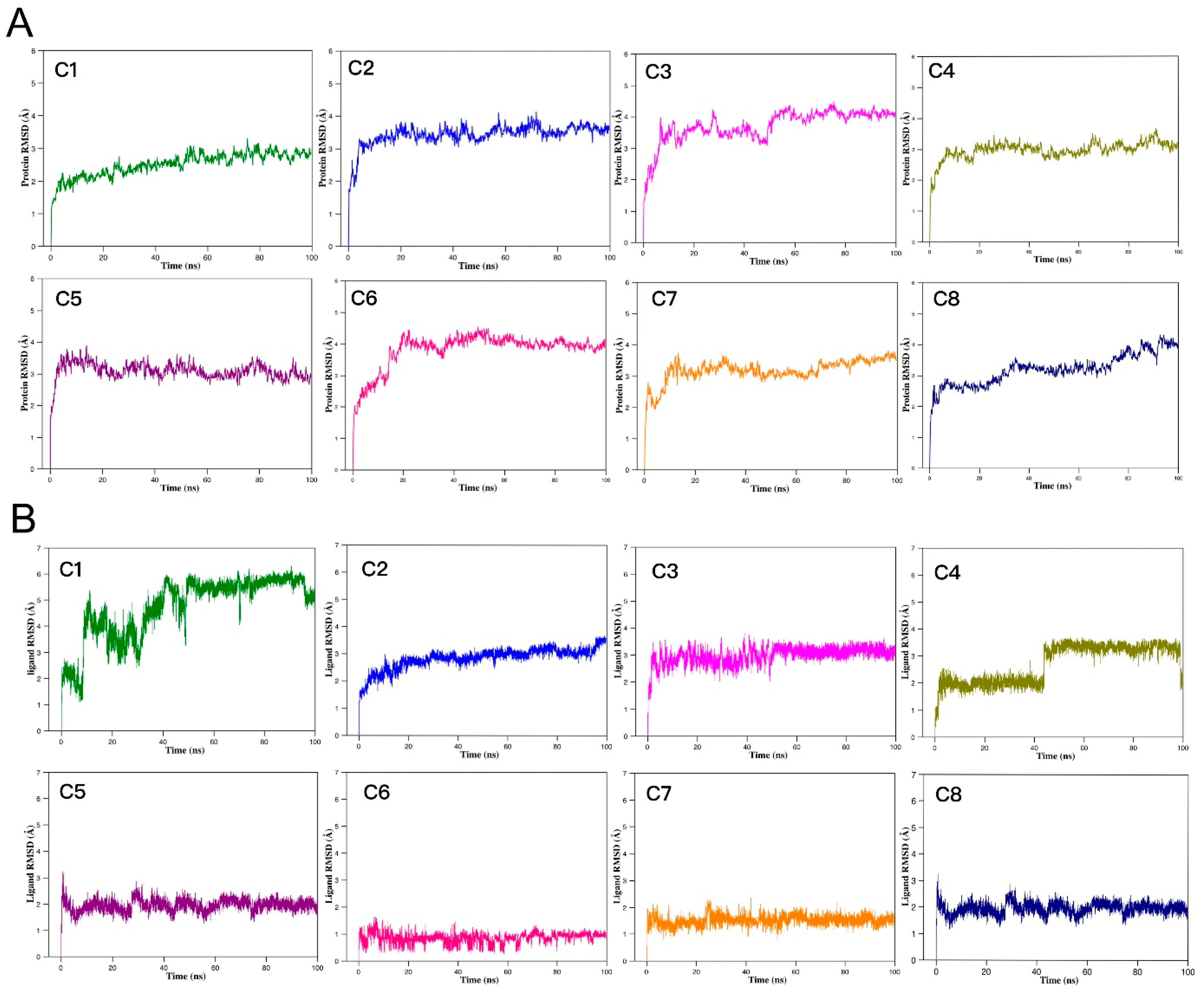
2.9. MD Simulations and Binding Free Energy Analysis
3. Discussion and Conclusions
4. Methods
4.1. Pan-Cancer Mutational Data Retrieval
4.2. Pan-Cancer GPR15 Expression Profile Analysis
4.3. Integrated Network Analysis of GPR15
4.4. Survival Analysis of GPR15 Expression
4.5. Gene Differential Expression Analysis
4.6. Commonly Upregulated Gene Set Identification and Annotation
4.7. 3D Structure Prediction and Validation of GPR15
4.8. Active Site Prediction
4.9. Screening of Potential Compounds Targeting GPR15
4.10. Molecular Docking
4.11. Molecular Dynamics (MD) Simulations
4.12. MD Trajectories Analysis
4.13. Binding Free Energy Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder urothelial carcinoma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell Carcinoma and endocervical adenocarcinoma |
| CHOL | Cholangiocarcinoma |
| COAD | Colon adenocarcinoma |
| DLBC | Diffuse large B-cell lymphoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma multiforme |
| HNSC | Head and neck squamous cell carcinoma |
| KICH | Kidney chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LAML | Acute myeloid leukemia |
| LGG | Lower grade glioma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| MESO | Mesothelioma |
| OV | Ovarian serous cystadenocarcinoma |
| PADD | Pancreatic adenocarcinoma |
| PCPG | Pheochromocytoma and Paraganglioma |
| PRAD | prostate adenocarcinoma |
| READ | Rectum adenocarcinoma |
| SARC | Sarcoma |
| SKCM | Skin cutaneous melanoma |
| STAD | Stomach adenocarcinoma |
| TGCT | Testicular germ cell tumors |
| THCA | Thyroid carcinoma |
| THYM | Thymoma |
| UCEC | Uterine corpus endometrial carcinoma |
| UCS | Uterine carcinosarcoma |
| UVM | Uveal Melanoma |
References
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.K.; Unutmaz, D.; KewalRamani, V.N.; Littman, D.R. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 1997, 388, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Wang, J.Y. G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. Int. J. Mol. Sci. 2016, 17, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Huang, S.; Peng, S.B. Overexpression of G protein-coupled receptors in cancer cells: Involvement in tumor progression. Int. J. Oncol. 2005, 27, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Gugger, M.; White, R.; Song, S.; Waser, B.; Cescato, R.; Riviere, P.; Reubi, J.C. GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis. Markers 2008, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Luo, R.; Piao, X. GPR56 and its related diseases. Prog. Mol. Biol. Transl. Sci. 2009, 89, 1–13. [Google Scholar]
- Kim, S.V.; Xiang, W.V.; Kwak, C.; Yang, Y.; Lin, X.W.; Ota, M.; Sarpel, U.; Rifkin, D.B.; Xu, R.; Littman, D.R. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 2013, 340, 1456–1459. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [Green Version]
- Heiber, M.; Marchese, A.; Nguyen, T.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. A novel human gene encoding a G-protein-coupled receptor (GPR15) is located on chromosome 3. Genomics 1996, 32, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Blaak, H.; Boers, P.H.M.; Gruters, R.A.; Schuitemaker, H.; Van Der Ende, M.E.; Osterhaus, A. CCR5, GPR15, and CXCR6 are major coreceptors of human immunodeficiency virus type 2 variants isolated from individuals with and without plasma viremia. J. Virol. 2005, 79, 1686–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.P.; Pan, J.; Dinh, T.T.; Hadeiba, H.; O’Hara, E., III; Ebtikar, A.; Hertweck, A.; Gokmen, M.R.; Lord, G.M.; Jenner, R.G.; et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat. Immunol. 2015, 16, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Seong, Y.; Lazarus, N.H.; Sutherland, L.; Habtezion, A.; Abramson, T.; He, X.S.; Greenberg, H.B.; Butcher, E.C. Trafficking receptor signatures define blood plasmablasts responding to tissue-specific immune challenge. JCI Insight 2017, 2, e90233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahl, K.; Sweere, J.; Pan, J.; Butcher, E. Orphan chemoattractant receptor GPR15 mediates dendritic epidermal T-cell recruitment to the skin. Eur. J. Immunol. 2014, 44, 2577–2581. [Google Scholar] [CrossRef] [Green Version]
- Habtezion, A.; Nguyen, L.P.; Hadeiba, H.; Butcher, E.C. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology 2016, 150, 340–354. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2017, 95, 325–332. [Google Scholar] [CrossRef]
- Haase, T.; Muller, C.; Krause, J.; Rothemeier, C.; Stenzig, J.; Kunze, S.; Waldenberger, M.; Munzel, T.; Pfeiffer, N.; Wild, P.S.; et al. Novel DNA Methylation Sites Influence GPR15 Expression in Relation to Smoking. Biomolecules 2018, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Koks, G.; Uudelepp, M.L.; Limbach, M.; Peterson, P.; Reimann, E.; Koks, S. Smoking-induced expression of the GPR15 gene indicates its potential role in chronic inflammatory pathologies. Am. J. Pathol. 2015, 185, 2898–2906. [Google Scholar] [CrossRef] [Green Version]
- Koks, S.; Koks, G. Activation of GPR15 and its involvement in the biological effects of smoking. Exp. Biol. Med. 2017, 242, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Ocón, B.; Pan, J.; Dinh, T.T.; Chen, W.; Ballet, R.; Bscheider, M.; Habtezion, A.; Tu, H.; Zabel, B.A.; Butcher, E.C. A Mucosal and Cutaneous Chemokine Ligand for the Lymphocyte Chemoattractant Receptor GPR15. Front. Immunol. 2017, 8, 1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Cheng, Y.; Zhang, H.; Liu, B.; Mo, X.; Li, T.; Li, L.; Cheng, X.; Zhang, L.; Ji, J.; et al. CSBF/C10orf99, a novel potential cytokine, inhibits colon cancer cell growth through inducing G1 arrest. Sci. Rep. 2014, 4, 6812. [Google Scholar] [CrossRef] [PubMed]
- Suply, T.; Hannedouche, S.; Carte, N.; Li, J.P.; Grosshans, B.; Schaefer, M.; Raad, L.; Beck, V.; Vidal, S.; Hiou-Feige, A.; et al. A natural ligand for the orphan receptor GPR15 modulates lymphocyte recruitment to epithelia. Sci. Signal. 2017, 10, eaal0180. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kaddi, C.D.; Wang, M.D. Pan-cancer analysis for studying cancer stage using protein and gene expression data. In Proceedings of the IEEE Engineering in Medicine and Biology Society, Orlando, FL, USA, 16–20 August 2016; pp. 2440–2443. [Google Scholar]
- Morris, L.G.; Riaz, N.; Desrichard, A.; Senbabaoglu, Y.; Hakimi, A.A.; Makarov, V.; Reis-Filho, J.S.; Chan, T.A. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016, 7, 10051–10063. [Google Scholar] [CrossRef] [Green Version]
- Ng, J.C.F.; Quist, J.; Grigoriadis, A.; Malim, M.H.; Fraternali, F. Pan-cancer transcriptomic analysis dissects immune and proliferative functions of APOBEC3 cytidine deaminases. Nucleic Acids Res. 2019, 47, 1178–1194. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. Cancer Genome Atlas Research Network. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar] [CrossRef]
- Singh, A.; Goel, N.; Yogita. Integrative Analysis of Multi-Genomic Data for Kidney Renal Cell Carcinoma. Interdiscip. Sci. 2019. [Google Scholar] [CrossRef]
- Chen, H.; Fu, W.; Wang, Z.; Wang, X.; Lei, T.; Zhu, F.; Li, D.; Chang, S.; Xu, L.; Hou, T. Reliability of Docking-Based Virtual Screening for GPCR Ligands with Homology Modeled Structures: A Case Study of the Angiotensin II Type I Receptor. ACS Chem. Neurosci. 2019, 10, 677–689. [Google Scholar] [CrossRef]
- Varano, F.; Catarzi, D.; Falsini, M.; Vincenzi, F.; Pasquini, S.; Varani, K.; Colotta, V. Identification of novel thiazolo[5,4-d]pyrimidine derivatives as human A1 and A2A adenosine receptor antagonists/inverse agonists. Bioorg. Med. Chem. 2018, 26, 3688–3695. [Google Scholar] [CrossRef]
- Cooke, R.M.; Brown, A.J.; Marshall, F.H.; Mason, J.S. Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discov. Today 2015, 20, 1355–1364. [Google Scholar] [CrossRef]
- Becker, O.M.; Shacham, S.; Marantz, Y.; Noiman, S. Modeling the 3D structure of GPCRs: Advances and application to drug discovery. Curr. Opin. Drug Discov. Dev. 2003, 6, 353–361. [Google Scholar]
- Becker, O.M.; Marantz, Y.; Shacham, S.; Inbal, B.; Heifetz, A.; Kalid, O.; Bar-Haim, S.; Warshaviak, D.; Fichman, M.; Noiman, S. G protein-coupled receptors: In silico drug discovery in 3D. Proc. Natl. Acad. Sci. USA 2004, 101, 11304–11309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, N.; Anand, P.; Yeturu, K. Structural Bioinformatics: Deriving Biological Insights from Protein Structures. Interdiscip. Sci. Comput. Life Sci. 2010, 2, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Gautam, D.; Nangraj, A.S.; Wei, D.Q.; Sahi, S. Protection of Primary Dopaminergic Midbrain Neurons Through Impact of Small Molecules Using Virtual Screening of GPR139 Supported by Molecular Dynamic Simulation and Systems Biology. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Mahajanakatti, A.B.; Murthy, G.; Sharma, N.; Skariyachan, S. Exploring Inhibitory Potential of Curcumin against Various Cancer Targets by in silico Virtual Screening. Interdiscip. Sci. Comput. Life Sci. 2014, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Wang, X.G.; Xiong, Y.; Kaushik, A.C.; Muhammad, J.; Khan, A.; Dai, H.; Wei, D.Q. New strategy for identifying potential natural HIV-1 non-nucleoside reverse transcriptase inhibitors against drug-resistance: An in silico study. J. Biomol. Struct. Dyn. 2019. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Takihara, Y.; Matsuda, Y.; Hara, J. Role of the beta isoform of 14-3-3 proteins in cellular proliferation and oncogenic transformation. Carcinogenesis 2000, 21, 2073–2077. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Razick, S.; Magklaras, G.; Donaldson, I.M. iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinf. 2008, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Tateno, H.; Matsushima, A.; Hiemori, K.; Onuma, Y.; Ito, Y.; Hasehira, K.; Nishimura, K.; Ohtaka, M.; Takayasu, S.; Nakanishi, M.; et al. Podocalyxin Is a Glycoprotein Ligand of the Human Pluripotent Stem Cell-Specific Probe rBC2LCN. Stem Cells Transl. Med. 2013, 2, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Hannenhalli, S.; Putt, M.E.; Gilmore, J.M.; Wang, J.W.; Parmacek, M.S.; Epstein, J.A.; Morrisey, E.E.; Margulies, K.B.; Cappola, T.P. Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation 2006, 114, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, S.G.; Yin, L.L.; Smuda, C.; Mahns, A.; Kolbe, L.; Hearing, V.J. Photobiological implications of melanin photoprotection after UVB-induced tanning of human skin but not UVA-induced tanning. Pigment Cell Melanoma Res. 2015, 28, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholtysik, R.; Kreuz, M.; Hummel, M.; Rosolowski, M.; Szczepanowski, M.; Klapper, W.; Loeffler, M.; Trumper, L.; Siebert, R.; Kuppers, R.; et al. Characterization of genomic imbalances in diffuse large B-cell lymphoma by detailed SNP-chip analysis. Int. J. Cancer 2015, 136, 1033–1042. [Google Scholar] [CrossRef]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Zhang, C.X.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; Cherezov, V.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2017, 46, D440–D446. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andradas, C.; Caffarel, M.M.; Perez-Gomez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2011, 30, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartwright, A.; Schmutz, C.; Askari, A.; Kuiper, J.H.; Middleton, J. Orphan receptor GPR15/BOB is up-regulated in rheumatoid arthritis. Cytokine 2014, 67, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 10001. [Google Scholar] [CrossRef] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.R.; Jurisica, I. Online Predicted Human Interaction Database. Bioinformatics 2005, 21, 2076–2082. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Ray, D.; Warde-Farley, D.; Grouios, C.; Morris, Q. GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9, S4. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R. The Reactome pathway knowledgebase. Nucleic Acids Res. 2013, 42, D472–D477. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using’ggplot2’, R Package Version 03; Available online: https://rpkgs.datanovia.com/survminer/index.html (accessed on 3 September 2019).
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Swinton, J. Vennerable: Venn and Euler Area-Proportional Diagrams, R Package Version 03; Available online: https://rdrr.io/rforge/Vennerable/ (accessed on 2 May 2019).
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. Clusterprofiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sanchez, R.; Sali, A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins 1997, 1, 50–58. [Google Scholar] [CrossRef]
- Rodrigues, J.P.; Levitt, M.; Chopra, G. KoBaMIN: A knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res. 2012, 40, W323–W328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.N. Scoring noncovalent protein-ligand interactions: A continuous differentiable function tuned to compute binding affinities. J. Comput.-Aided Mol. Des. 1996, 10, 427–440. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R. Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Score | Network Group | Network Resource |
|---|---|---|---|
| YWHAB | 0.22097643 | Physical Interactions | BioGRID-small-scale-studies [40] |
| YWHAB | 0.14404532 | Physical Interactions | IREF-INTACT [41] |
| TACR1 | 0.038140558 | Co-expression | Tateno-Hirabayashi 2013 [42] |
| TAS2R9 | 0.034424774 | Co-expression | Hannenhalli-Cappola 2006 [43] |
| SPDYE4 | 0.031733938 | Co-expression | Coelho-Hearing 2015 [44] |
| GPR182 | 0.029303862 | Co-expression | Scholtysik-Kuppers 2015 [45] |
| Pathway ID | Pathway Name | p-Value | Entities Found |
|---|---|---|---|
| R-HSA-450385 | Butyrate Response Factor 1 (BRF1) binds and destabilizes mRNA | 0.00231 | YWHAB;EXOSC1 |
| R-HSA-450513 | Tristetraprolin (TTP, ZFP36) binds and destabilizes mRNA | 0.00231 | YWHAB;EXOSC1 |
| R-HSA-525793 | Myogenesis | 0.00636 | MYF6;MEF2D |
| R-HSA-375170 | CDO in myogenesis | 0.00636 | MYF6;MEF2D |
| R-HSA-388396 | GPCR downstream signaling | 0.00967 | GPR15 |
| Cancer | Cox Coefficient | p-Value | Rank |
|---|---|---|---|
| STAD | 0.27 | 0.002 | 269 |
| HNSC | −0.205 | 0.006 | 707 |
| LUAD | −0.161 | 0.039 | 3711 |
| COAD | −0.159 | 0.150 | 4299 |
| READ | −0.328 | 0.160 | 2696 |
| LUSC | 0.07 | 0.330 | 6956 |
| KIRC | −0.059 | 0.480 | 13,174 |
| LAML | 0.078 | 0.510 | 9516 |
| ESCA | 0.021 | 0.880 | 14,833 |
| ID | Description | p-Adjust | Category |
|---|---|---|---|
| GO:0006958 | complement activation, classical pathway | 2.78 × 10−120 | BP |
| GO:0002455 | humoral immune response mediated by circulating immunoglobulin | 3.03 × 10−117 | BP |
| GO:0006956 | complement activation | 2.21× 10−112 | BP |
| GO:0072376 | protein activation cascade | 1.16 × 10−107 | BP |
| GO:0016064 | immunoglobulin mediated immune response | 1.36 × 10−104 | BP |
| GO:0019724 | B cell mediated immunity | 1.66 × 10−104 | BP |
| GO:0002429 | immune response-activating cell surface receptor signaling pathway | 3.22 × 10−91 | BP |
| GO:0006959 | humoral immune response | 4.60 × 10−91 | BP |
| GO:0002768 | immune response-regulating cell surface receptor signaling pathway | 8.99 × 10−94 | BP |
| GO:0002460 | adaptive immune response based on somatic recombination of immune receptors | 1.21 × 10−91 | BP |
| GO:0019814 | immunoglobulin complex | 1.32 × 10−80 | CC |
| GO:0042571 | immunoglobulin complex, circulating | 3.13 × 10−77 | CC |
| GO:0009897 | external side of plasma membrane | 1.50 × 10−44 | CC |
| GO:0072562 | blood microparticle | 2.80 × 10−18 | CC |
| GO:0098802 | plasma membrane receptor complex | 0.513620478 | CC |
| GO:0042101 | T cell receptor complex | 0.513620478 | CC |
| GO:0008180 | COP9 signalosome | 0.721923256 | CC |
| GO:0043235 | receptor complex | 0.721923256 | CC |
| GO:0000788 | nuclear nucleosome | 0.721923256 | CC |
| GO:0005771 | multivesicular body | 0.754761177 | MF |
| GO:0003823 | antigen binding | 2.41 × 10−159 | MF |
| GO:0034987 | immunoglobulin receptor binding | 2.28 × 10−71 | MF |
| GO:0004252 | serine-type endopeptidase activity | 2.71 × 10−46 | MF |
| GO:0008236 | serine-type peptidase activity | 9.31 × 10−45 | MF |
| GO:0017171 | serine hydrolase activity | 1.40 × 10−44 | MF |
| GO:0005068 | transmembrane receptor protein tyrosine kinase adaptor activity | 0.03233891 | MF |
| GO:0042834 | peptidoglycan binding | 0.055957242 | MF |
| GO:0031210 | phosphatidylcholine binding | 0.100244352 | MF |
| GO:0050997 | quaternary ammonium group binding | 0.100244352 | MF |
| GO:0035591 | signaling adaptor activity | 0.102967022 | MF |
| Compound No. | Molecular Formula | Weight (g/mol) | Docking Score | Noncovalent Interactions | Residues |
|---|---|---|---|---|---|
| C1 | C38H58O2N2 | 576.91 | −11.63 | 2 Pi–pi, 2 H–bond | TRP89, ASP91 |
| C2 | C60H55O8N1 | 918.09 | −11.15 | 1 Pi–pi, 2 Pi–cation, 1 H–Bond | LYS180, ARG172, TRP195, LYS261 |
| C3 | C38H41O7N3 | 653.77 | −10.79 | 2 H–Bond | CYS183, ARG172 |
| C4 | C21H28O4N2S | 404.53 | −10.28 | 1 Pi–pi, 2 H–bond | TRP89, SER109, LYS180 |
| C5 | C34H47O6N3 | 593.76 | −10.11 | 1 Pi–pi, 1 Salt–bridge | PHE257, LYS261 |
| C6 | C27H46O3 | 418.66 | −8.72 | 2 H–bond | ARG172, LYS180 |
| C7 | C20H24O4 | 328.41 | −8.3 | 2 Pi–pi, 1 H–bond | TRP89, TYR182, LYS180 |
| C8 | C22H27O5N5S | 473.55 | −8.29 | 1 Salt–bridge,1 Pi–pi | LYS261, TRP89 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, X.; Xiong, Y.; Li, C.-D.; Xu, Q.; Shen, L.; Chandra Kaushik, A.; Wei, D.-Q. An Integrated Pan-Cancer Analysis and Structure-Based Virtual Screening of GPR15. Int. J. Mol. Sci. 2019, 20, 6226. https://doi.org/10.3390/ijms20246226
Wang Y, Wang X, Xiong Y, Li C-D, Xu Q, Shen L, Chandra Kaushik A, Wei D-Q. An Integrated Pan-Cancer Analysis and Structure-Based Virtual Screening of GPR15. International Journal of Molecular Sciences. 2019; 20(24):6226. https://doi.org/10.3390/ijms20246226
Chicago/Turabian StyleWang, Yanjing, Xiangeng Wang, Yi Xiong, Cheng-Dong Li, Qin Xu, Lu Shen, Aman Chandra Kaushik, and Dong-Qing Wei. 2019. "An Integrated Pan-Cancer Analysis and Structure-Based Virtual Screening of GPR15" International Journal of Molecular Sciences 20, no. 24: 6226. https://doi.org/10.3390/ijms20246226
APA StyleWang, Y., Wang, X., Xiong, Y., Li, C. -D., Xu, Q., Shen, L., Chandra Kaushik, A., & Wei, D. -Q. (2019). An Integrated Pan-Cancer Analysis and Structure-Based Virtual Screening of GPR15. International Journal of Molecular Sciences, 20(24), 6226. https://doi.org/10.3390/ijms20246226