A Novel DES L115F Mutation Identified by Whole Exome Sequencing is Associated with Inherited Cardiac Conduction Disease

 ,
, 
Abstract
:1. Introduction
2. Results
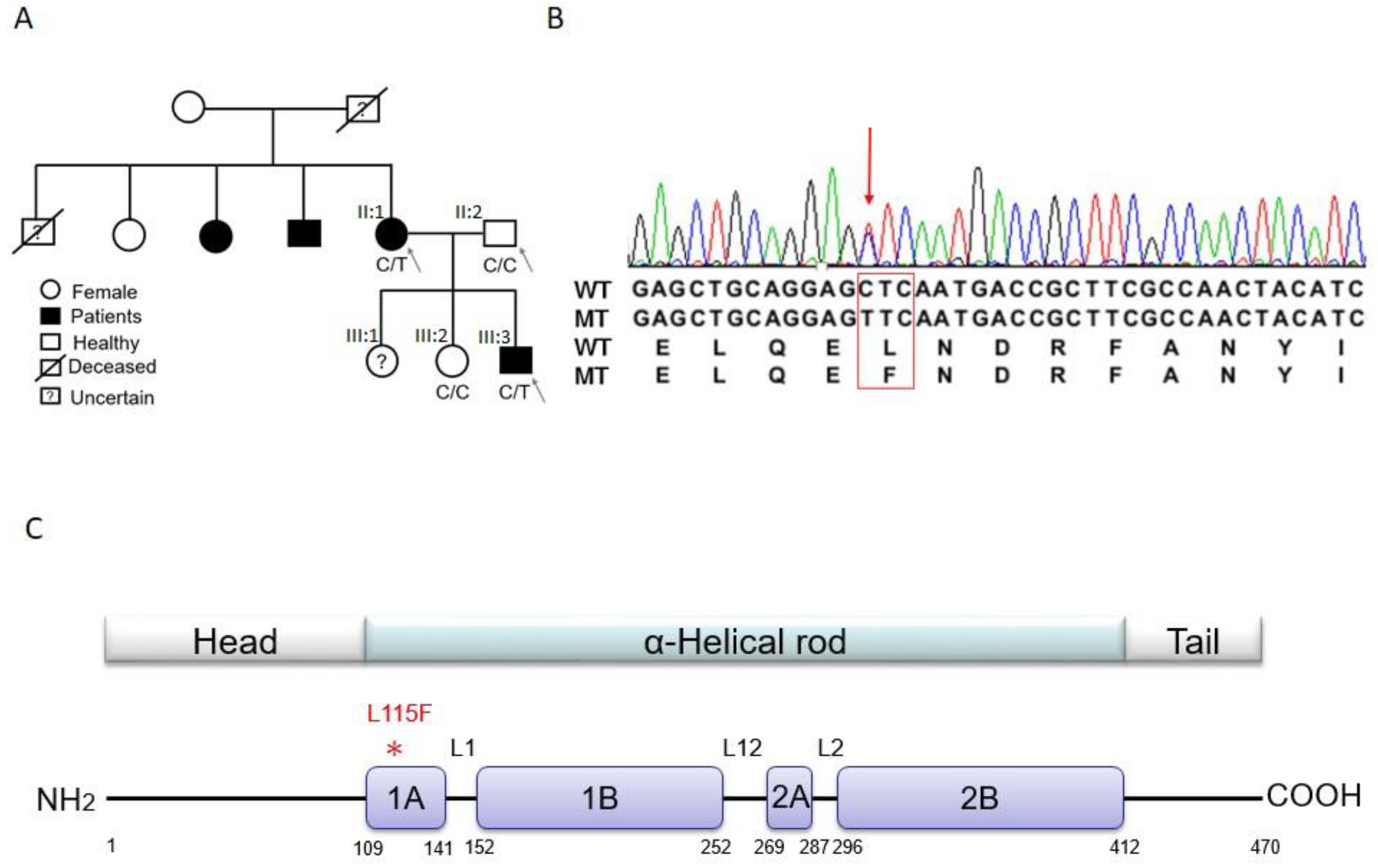
2.1. Trio-based Exome Sequencing and Variant Filtration and Prioritization
2.2. Validation of Candidate Genes
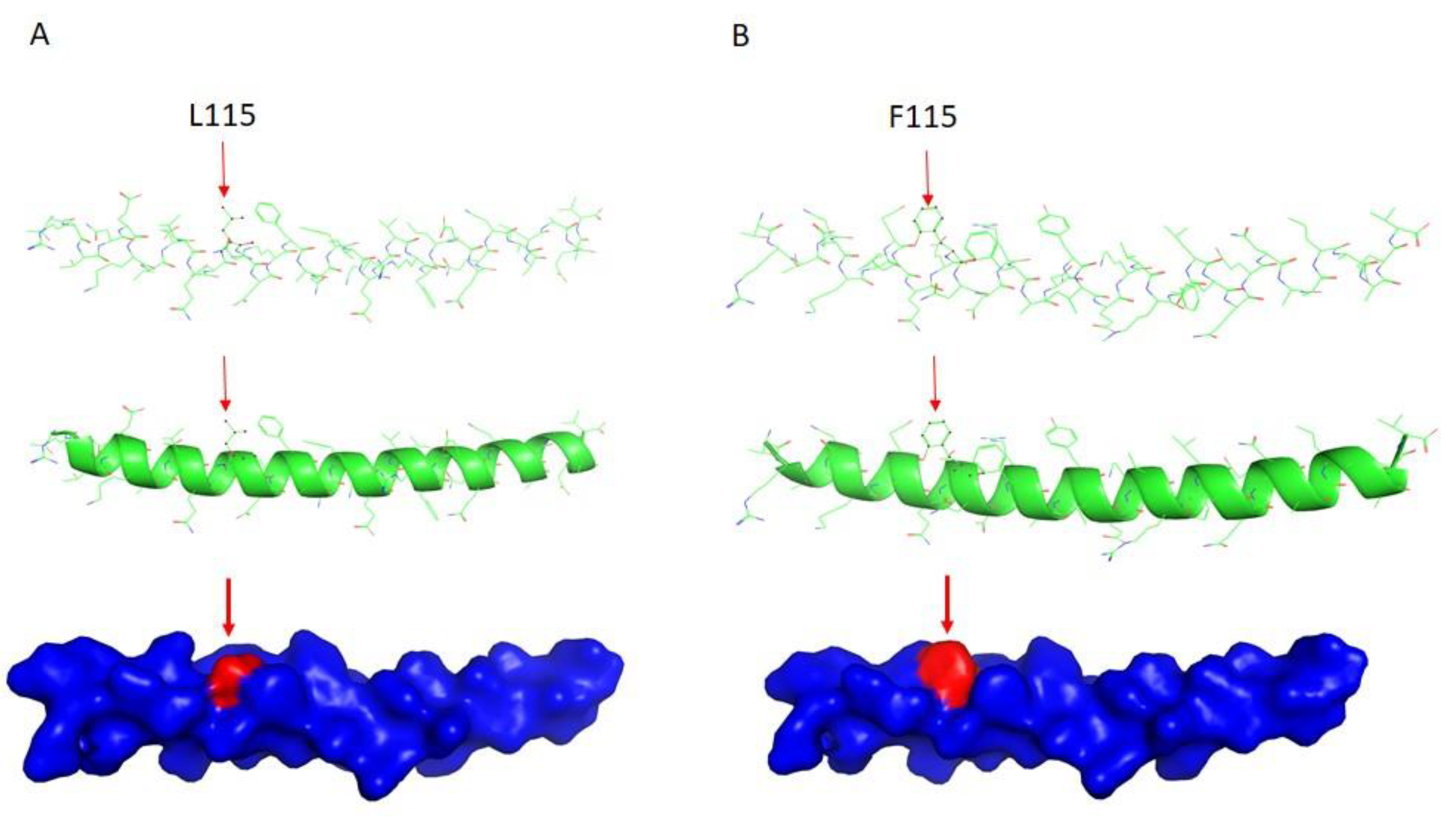
2.3. DES Wild-Type/Mutation Structure Modeling
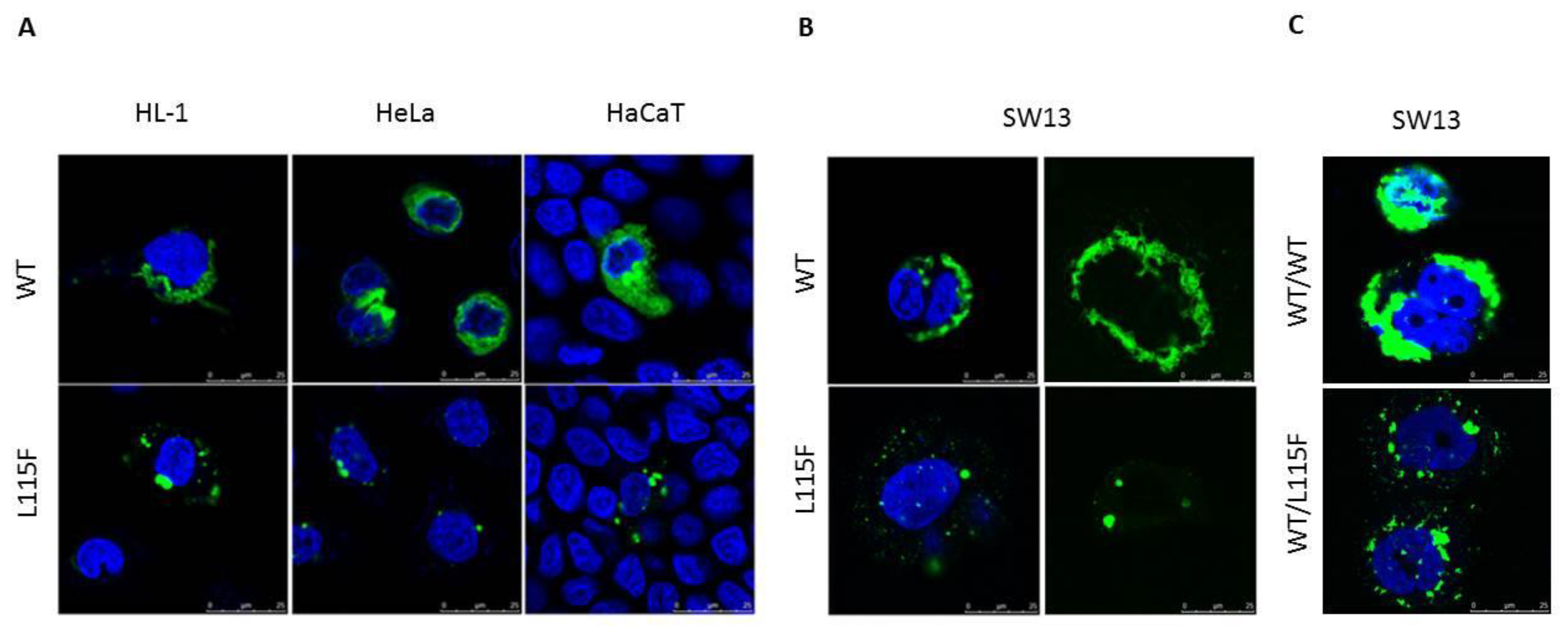
2.4. Cytoplasmic Aggregate Formation Caused by the Desmin Mutant in Transfected Cells
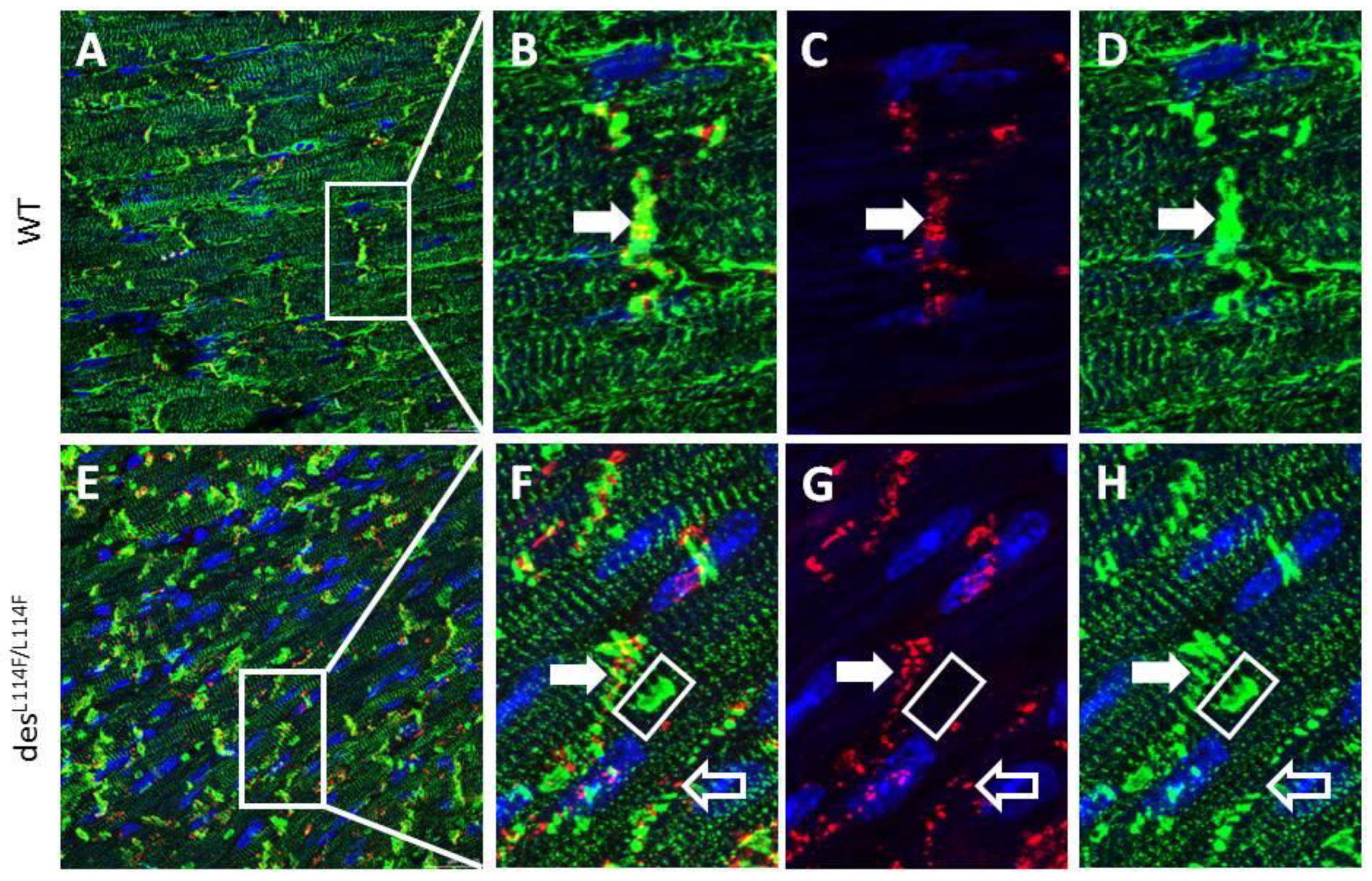
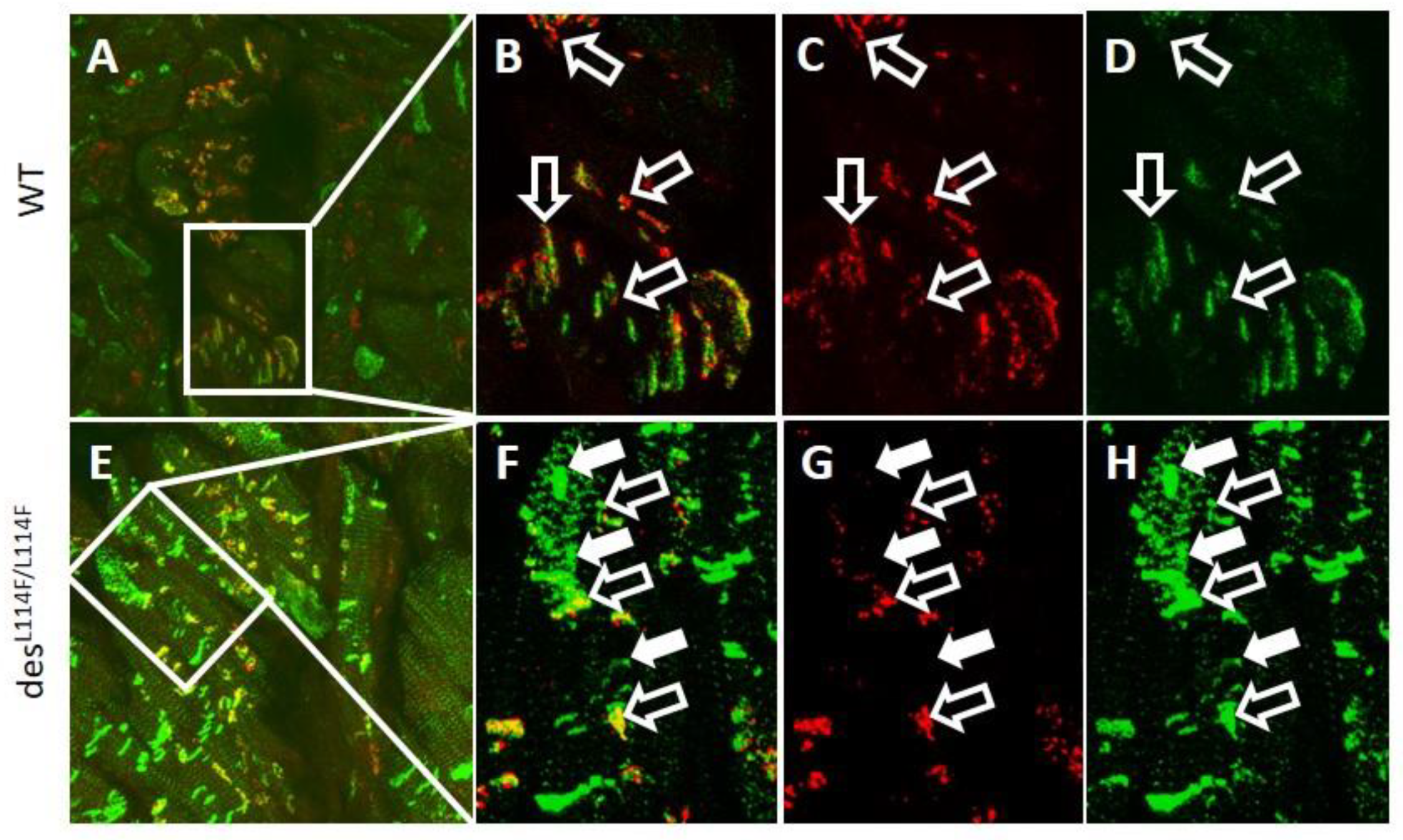
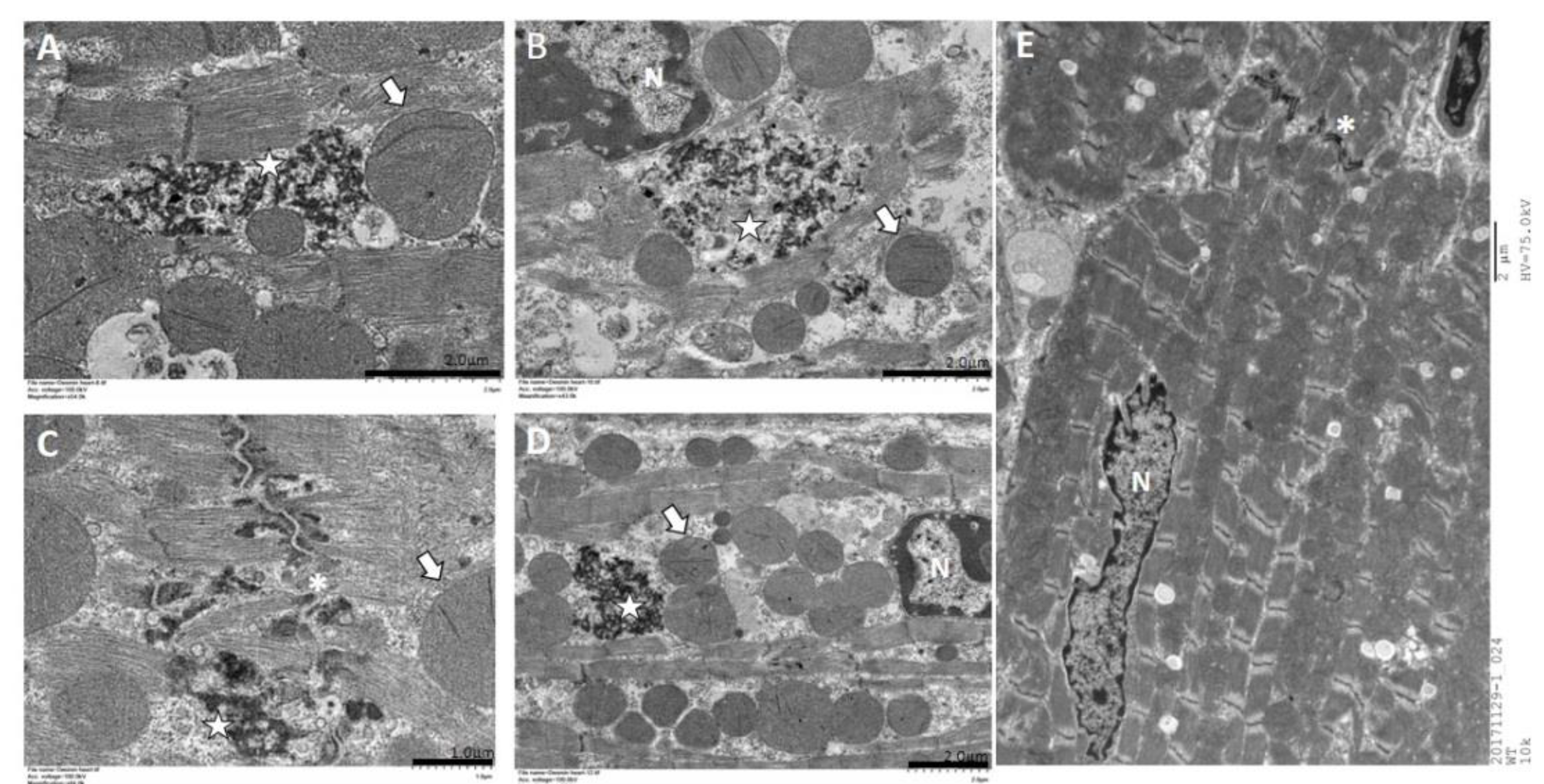
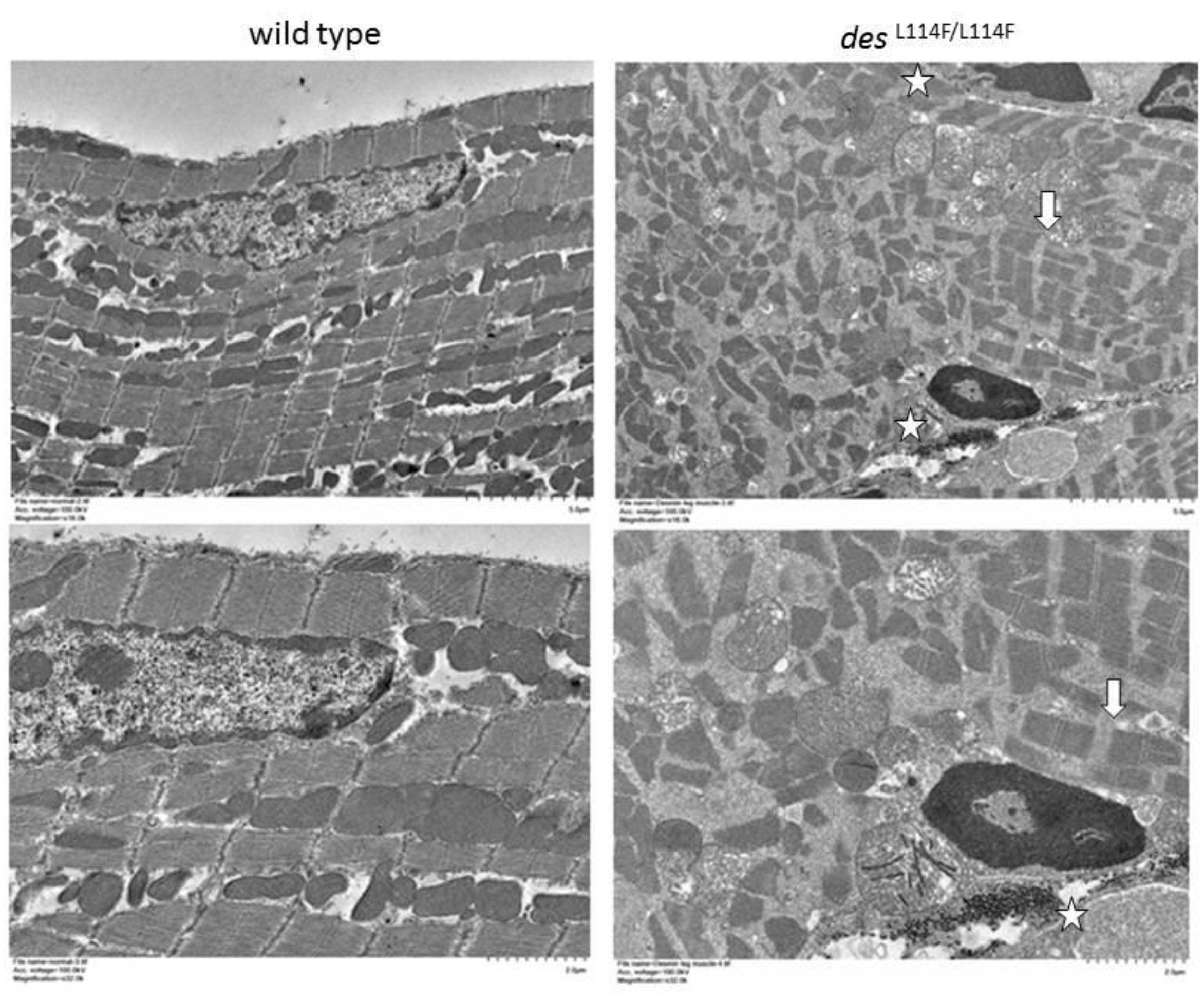
2.5. Immunohistochemistry and Electron Microscopy in des L114F Knock-in Mice
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Study Population
4.3. Exome Sequencing and Variant Calling
4.4. Mutation Validation
4.5. DES Wild-Type/Mutation Structure Modeling
4.6. Cell Culture
4.7. Expression Analysis
4.8. Generation of desL114F Knock-in Mice Using CRISPR/CAS9
4.9. Immunohistochemistry
4.10. Transmission Electron Microscopy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wolf, C.M.; Berul, C.I. Inherited conduction system abnormalities--one group of diseases, many genes. J. Cardiovasc. Electrophysiol. 2006, 17, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Smits, J.P.; Veldkamp, M.W.; Wilde, A.A. Mechanisms of inherited cardiac conduction disease. Europace 2005, 7, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Fishman, G.I. The cardiac conduction system. Circulation 2011, 123, 904–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.C.; Yeh, Y.H.; Hsieh, W.P.; Kuo, C.T.; Wang, W.C.; Chu, C.H.; Hung, C.L.; Cheng, C.Y.; Tsai, H.Y.; Lee, J.L.; et al. Whole-exome sequencing to identify a novel LMNA gene mutation associated with inherited cardiac conduction disease. PLoS ONE 2013, 8, e83322. [Google Scholar] [CrossRef]
- Chernyatina, A.A.; Nicolet, S.; Aebi, U.; Herrmann, H.; Strelkov, S.V. Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 13620–13625. [Google Scholar] [CrossRef] [Green Version]
- Strelkov, S.V.; Herrmann, H.; Geisler, N.; Wedig, T.; Zimbelmann, R.; Aebi, U.; Burkhard, P. Conserved segments 1A and 2B of the intermediate filament dimer: Their atomic structures and role in filament assembly. EMBO J. 2002, 21, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Meier, M.; Padilla, G.P.; Herrmann, H.; Wedig, T.; Hergt, M.; Patel, T.R.; Stetefeld, J.; Aebi, U.; Burkhard, P. Vimentin coil 1A-A molecular switch involved in the initiation of filament elongation. J. Mol. Biol. 2009, 390, 245–261. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Šarić, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, preventsintercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, I.; Dieding, M.; Klauke, B.; Brodehl, A.; Gaertner-Rommel, A.; Walhorn, V.; Gummert, J.; Schulz, U.; Paluszkiewicz, L.; Anselmetti, D.; et al. A novel desmin (DES) indel mutation causes severe atypical cardiomyopathy in combination with atrioventricular block and skeletal myopathy. Mol. Genet. Genomic. Med. 2018, 6, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Colucci-Guyon, E.; Pincon-Raymond, M.; Mericskay, M.; Pournin, S.; Paulin, D.; Babinet, C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev. Biol. 1996, 175, 362–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degenerationin mice lacking desmin. J. Cell. Bio. 1996, 134, 1255–1270. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Goldfarb, L.G.; Dalakas, M.C. Tragedy in a heartbeat: Malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Invest. 2009, 119, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef]
- Kostera-Pruszczyk, A.; Pruszczyk, P.; Kaminska, A.; Lee, H.S.; Goldfarb, L.G. Diversity of cardiomyopathy phenotypes caused by mutations in desmin. Int. J. Cardiol. 2007, 131, 146–147. [Google Scholar] [CrossRef]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Vernengo, L.; Chourbagi, O.; Panuncio, A.; Lilienbaum, A.; Batonnet-Pichon, S.; Bruston, F.; Rodrigues-Lima, F.; Mesa, R.; Pizzarossa, C.; Demay, L.; et al. Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul. Disord. 2010, 20, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Price, M.G. Molecular analysis of intermediate filament cytoskeleton—a putative load-bearing structure. Am. J. Physiol. 1984, 246, H566–H572. [Google Scholar] [CrossRef] [PubMed]
- Gard, J.J.; Yamada, K.; Green, K.G.; Eloff, B.C.; Rosenbaum, D.S.; Wang, X.; Robbins, J.; Schuessler, R.B.; Yamada, K.A.; Saffitz, J.E. Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc. Res. 2005, 67, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Patel, V.V.; Radice, G.L. Dysregulation of cell adhesion proteins and cardiac arrhythmogenesis. Clin. Med. Res. 2006, 4, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Clemen, C.S.; Stöckigt, F.; Strucksberg, K.H.; Chevessier, F.; Winter, L.; Schütz, J.; Bauer, R.; Thorweihe, J.M.; Wenzel, D.; Schlötzer-Schrehardt, U.; et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015, 129, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Winter, L.; Unger, A.; Berwanger, C.; Spörrer, M.; Türk, M.; Chevessier, F.; Strucksberg, K.H.; Schlötzer-Schrehardt, U.; Wittig, I.; Goldmann, W.H.; et al. Imbalances in protein homeostasis caused by mutant desmin. Neuropathol. Appl. Neurobiol. 2019, 45, 476–494. [Google Scholar] [CrossRef]
- Sung, Y.H.; Jin, Y.; Kim, S.; Lee, H.W. Generation of knockout mice using engineered nucleases. Methods 2014, 69, 85–93. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex | Conduction Defect | Paf | Age at Echo | LV EF, % | LA, mm | LV, mm | Age at PPM | |
|---|---|---|---|---|---|---|---|---|
| II:1 | F | SND, 3rd-degree AVB | yes | 55 | 73 | 47 | 46 | 55 |
| II:2 | M | No | No | 65 | 80 | 37 | 50 | |
| III:2 | F | No | No | |||||
| III:3 | M | SND, 1st-degree AVB | yes | 35 | 64 | 38 | 43 | 35 |
| Steps | Residual SNVs & InDels | Percentage (%) | |
|---|---|---|---|
| TVC filtered variants | 39,229 | 100 | |
| 1 | exonic or splicing site variants | 15,206 | 38.76 |
| 2 | selected heterozygous variants | 8935 | 22.78 |
| 3 | excluded synonymous SNVs | 4279 | 10.91 |
| 4 | excluded the variants with minor allele frequencies >1% in the dbSNP (version 142) or in the 5000Exomes Project | 230 | 0.59 |
| 5 | excluded benign variants (SIFT < 0.05 or Polyphen2 > 0.85) | 226 | 0.58 |
| 6 | variants coexisting within the affected mother and affected son | 68 | 0.17 |
| 7 | excluded the variants coexisting within the affected mother, affected son, and unaffected father | 56 | 0.14 |
| 8 | variants within hereditary cardiovascular disease-related genes | 5 | 0.01 |
| Gene Name | Primer Sequence | PCR Size (bp) | RE | Allele | Amino Acid Change |
|---|---|---|---|---|---|
| DES | (F)-5’-GTCCCGCGTGTACCAGGTGTC-3’, (R)-5’-CCAAGAAAACTCCTGTGCAAGATG-3’ | 646 | SacI | C>T | L115F |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, L.-A.; Ko, Y.-S.; Yeh, Y.-H.; Chang, C.-J.; Chan, Y.-H.; Kuo, C.-T.; Tsai, H.-Y.; Chang, G.-J. A Novel DES L115F Mutation Identified by Whole Exome Sequencing is Associated with Inherited Cardiac Conduction Disease. Int. J. Mol. Sci. 2019, 20, 6227. https://doi.org/10.3390/ijms20246227
Hsu L-A, Ko Y-S, Yeh Y-H, Chang C-J, Chan Y-H, Kuo C-T, Tsai H-Y, Chang G-J. A Novel DES L115F Mutation Identified by Whole Exome Sequencing is Associated with Inherited Cardiac Conduction Disease. International Journal of Molecular Sciences. 2019; 20(24):6227. https://doi.org/10.3390/ijms20246227
Chicago/Turabian StyleHsu, Lung-An, Yu-Shien Ko, Yung-Hsin Yeh, Chi-Jen Chang, Yi-Hsin Chan, Chi-Tai Kuo, Hsin-Yi Tsai, and Gwo-Jyh Chang. 2019. "A Novel DES L115F Mutation Identified by Whole Exome Sequencing is Associated with Inherited Cardiac Conduction Disease" International Journal of Molecular Sciences 20, no. 24: 6227. https://doi.org/10.3390/ijms20246227
APA StyleHsu, L. -A., Ko, Y. -S., Yeh, Y. -H., Chang, C. -J., Chan, Y. -H., Kuo, C. -T., Tsai, H. -Y., & Chang, G. -J. (2019). A Novel DES L115F Mutation Identified by Whole Exome Sequencing is Associated with Inherited Cardiac Conduction Disease. International Journal of Molecular Sciences, 20(24), 6227. https://doi.org/10.3390/ijms20246227