Fingolimod Plays Role in Attenuation of Myocardial Injury Related to Experimental Model of Cardiac Arrest and Extracorporeal Life Support Resuscitation

 , ,
, , 
Abstract
:
1. Introduction
2. Methods
2.1. Study Design and Setting
2.2. Animals
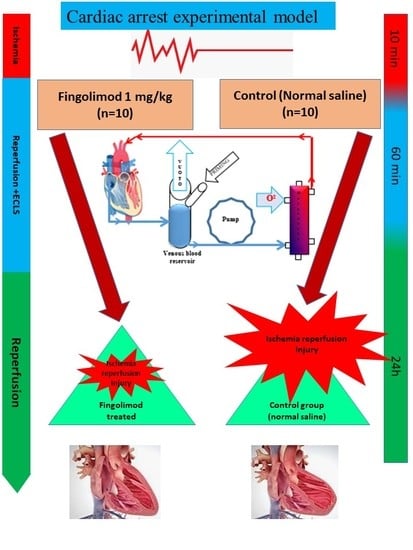
2.3. Experimental Design
2.4. Hemodynamic Analysis
2.5. Analysis of Serum Inflammatory Mediators and Biomarkers
2.6. Quantification of Oxidative Stress
2.7. Immunoblotting Analysis
2.8. Immunohistochemical Staining
2.9. High-Energy Phosphates
2.10. TUNEL Assay
2.11. Histology and Interstitial Fibrosis Determination
2.12. Statistical Analysis
3. Results
3.1. Left Ventricular Function
3.2. Serum Levels of Inflammatory Mediators and Cardiac Markers
3.3. Effect of Fingolimod on Nitro-Oxidative Stress
3.4. Bcl-2 and Bax Signaling Pathways
3.5. Effect of Fingolimod on Erk1/2 and Akt1/2 Signaling Pathways
3.6. High-Energy Phosphates
3.7. Effect of Fingolimod on Apoptosis
3.8. Effect of Fingolimod on Collagen Deposition and Neutrophil Infiltration
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jalife, J. The tornadoes of sudden cardiac arrest. Nature 2018, 555, 597–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinier, K.; Nichols, G.A.; Huertas-Vazquez, A.; Uy-Evanado, A.; Teodorescu, C.; Stecker, E.C.; Gunson, K.; Jui, J.; Chugh, S.S. Distinctive Clinical Profile of Blacks Versus Whites Presenting With Sudden Cardiac Arrest. Circulation 2015, 132, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stub, D.; Bernard, S.; Duffy, S.J.; Kaye Stub, D.M. Post cardiac arrest syndrome: A review of therapeutic strategies. Circulation 2011, 123, 1428–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongardon, N.; Dumas, F.; Ricome, S.; Grimaldi, D.; Hissem, T.; Pène, F.; Cariou, A. Postcardiac arrest syndrome: From immediate resuscitation to long-term outcome. Ann. Intensive Care 2011, 1, 45. [Google Scholar] [CrossRef] [Green Version]
- Nichol, G.; Karmy-Jones, R.; Salerno, C.; Cantore, L.; Becker, L. Systematic review of percutaneous cardiopulmonary bypass for cardiac arrest or cardiogenic shock states. Resuscitation 2006, 70, 381–394. [Google Scholar] [CrossRef]
- Chun, J.; Hla, T.; Lynch, K.R.; Spiegel, S.; Moolenaar, W.H. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid Receptor Nomenclature. Pharmacol. Rev. 2010, 62, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid receptors: Signaling and biology. Ann. Rev. Biochem. 2004, 73, 321–354. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. The J. of Cell Biol. 1991, 114, 155–167. [Google Scholar] [CrossRef]
- Hannun, Y.A. Functions of Ceramide in Coordinating Cellular Responses to Stress. Science 1996, 274, 1855–1859. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Luberto, C. Ceramide and the eukaryotic stress response. Biochem. Soc. Trans. 1997, 25, 1171–1175. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Cuvillier, O.; Edsall, L.C.; Kohama, T.; Menzeleev, R.; Olah, Z.; Olivera, A.; Pirianov, G.; Thomas, D.M.; Tu, Z.; et al. Sphingosine-1-phosphate in cell growth and cell death. Ann. N. Y. Acad. Sci. 1998, 845, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Linardi, D.; Muhammad, N.; Chiamulera, C.; Fumagalli, G.; Biagio, L.S.; Gebrie, M.A.; Aslam, M.; Luciani, G.B.; Faggian, G.; et al. Sphingosine 1-Phosphate Receptor Modulator Fingolimod (FTY720) Attenuates Myocardial Fibrosis in Post-heterotopic Heart Transplantation. Front. Pharmacol. 2017, 8, 645. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Linardi, D.; Decimo, I.; Mehboob, R.; Gebrie, M.A.; Innamorati, G.; Luciani, G.B.; Faggian, G.; Rungatscher, A. Characterization and Expression of Sphingosine 1-Phosphate Receptors in Human and Rat Heart. Front. Pharmacol. 2017, 8, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karliner, J.S.; Honbo, N.; Summers, K.; Gray, M.O.; Goetzl, E.J. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J. Mol. Cell Cardiol. 2001, 33, 1713–1717. [Google Scholar] [CrossRef]
- Jin, Z.Q.; Zhou, H.Z.; Zhu, P.; Honbo, N.; Mochly-Rosen, D.; Messing, R.O.; Goetzl, E.J.; Karliner, J.S.; Gray, M.O. Cardioprotection mediated by sphingosine-1-phosphate and ganglioside GM-1 in wild-type and PKC epsilon knockout mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1970–H1977. [Google Scholar] [CrossRef] [Green Version]
- Lecour, S.; Smith, R.M.; Woodward, B.; Opie, L.H.; Rochette, L.; Sack, M.N. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J. Mol. Cell Cardiol. 2002, 34, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Santos-Gallego, C.G.; Vahl, T.P.; Goliasch, G.; Picatoste, B.; Arias, T.; Ishikawa, K.; Njerve, I.U.; Sanz, J.; Narula, J.; Sengupta, P.P.; et al. Sphingosine-1-Phosphate Receptor Agonist Fingolimod Increases Myocardial Salvage and Decreases Adverse Postinfarction Left Ventricular Remodeling in a Porcine Model of Ischemia/Reperfusion. Circulation 2016, 133, 954–966. [Google Scholar] [CrossRef]
- Gorshkova, I.A.; Wang, H.; Orbelyan, G.A.; Goya, J.; Natarajan, V.; Beiser, D.G.; Vanden Hoek, T.L.; Berdyshev, E.V. Inhibition of sphingosine-1-phosphate lyase rescues sphingosine kinase-1-knockout phenotype following murine cardiac arrest. Life Sci. 2013, 93, 359–366. [Google Scholar] [CrossRef]
- Giani, J.F.; Gironacci, M.M.; Muñoz, M.C.; Peña, C.; Turyn, D.; Dominici, F.P. Angiotensin-(1 7) stimulates the phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: Role of the AT1 and Mas receptors. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1154–H1163. [Google Scholar] [CrossRef] [Green Version]
- Hallstrom, S.; Gasser, H.; Neumayer, C.; Fügl, A.; Nanobashvili, J.; Jakubowski, A.; Huk, I.; Schlag, G.; Malinski, T. S-nitroso human serum albumin treatment reduces ischemia/reperfusion injury in skeletal muscle via nitric oxide release. Circulation 2002, 105, 3032–3038. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Mehmood, A.; Linardi, D.; Sadiq, S.; Tessari, M.; Meo, S.A.; Rehman, R.; Hajjar, W.M.; Muhammad, N.; Iqbal, M.P.; et al. Cardioprotective Effects of Sphingosine-1-Phosphate Receptor Immunomodulator FTY720 in a Clinically Relevant Model of Cardioplegic Arrest and Cardiopulmonary Bypass. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giani, J.F.; Muñoz, M.C.; Mayer, M.A.; Veiras, L.C.; Arranz, C.; Taira, C.A.; Turyn, D.; Toblli, J.E.; Dominici, F.P. Angiotensin-(1-7) improves cardiac remodeling and inhibits growth-promoting pathways in the heart of fructose-fed rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1003–H1013. [Google Scholar] [CrossRef] [Green Version]
- Symons, J.A.; Myles, P.S. Myocardial protection with volatile anaesthetic agents during coronary artery bypass surgery: A meta-analysis. Br. J. Anaesth. 2006, 97, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Beattie, W.S. The effects of volatile anesthetics on cardiac ischemic complications and mortality in CABG: A meta-analysis. Can. J. Anaesth. 2006, 53, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, R.; Foëx, P. Myocardial protection by anesthetic agents against ischemia-reperfusion injury: An update for anesthesiologists. Can. J. Anaesth. 2002, 49, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Avkiran, M.; Marber, M.S. Na(+)/H(+) exchange inhibitors for cardioprotective therapy: Progress, problems and prospects. J. Am. Coll. Cardiol. 2002, 39, 747–753. [Google Scholar] [CrossRef]
- Pan, W.; Pintar, T.; Anton, J.; Lee, V.V.; Vaughn, W.K.; Collard, C.D. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004, 110, II45–II49. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.T.; LaFaro, R.J.; Reed, G.E. Pretreatment of human myocardium with adenosine during open heart surgery. J. Card. Surg. 1995, 10, 665–676. [Google Scholar] [CrossRef]
- Lutz, J.; Thürmel, K.; Heemann, U. Anti-inflammatory treatment strategies for ischemia/reperfusion injury in transplantation. J. Inflamm. 2010, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Brewster, B.D.; Rouch, J.D.; Wang, M.; Meldrum, D.R. Toll-like receptor 4 ablation improves stem cell survival after hypoxic injury. J. Surg. Res. 2012, 177, 330–333. [Google Scholar] [CrossRef]
- Bader, A.M.; Brodarac, A.; Klose, K.; Bieback, K.; Choi, Y.H.; Kurtz, A.; Stamm, C. Mechanisms of paracrine cardioprotection by cord blood mesenchymal stromal cells. Eur. J. Cardiothorac Surg. 2014, 45, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Kim, H.W.; Gong, M.; Wang, J.; Millard, R.W.; Wang, Y.; Ashraf, M.; Xu, M. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int. J. Cardiol. 2015, 182, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Means, C.K.; Xiao, C.Y.; Li, Z.; Zhang, T.; Omens, J.H.; Ishii, I.; Chun, J.; Brown, J.H. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2944–H2951. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.Q.; Zhang, J.; Huang, Y.; Hoover, H.E.; Vessey, D.A.; Karliner, J.S. A sphingosine kinase 1 mutation sensitizes the myocardium to ischemia/reperfusion injury. Cardiovasc. Res. 2007, 76, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Paillard, M.; Price, M.; Chen, Q.; Teixeira, G.; Spiegel, S.; Lesnefsky, E.J. A novel role for mitochondrial sphingosine-1-phosphate produced by sphingosine kinase-2 in PTP-mediated cell survival during cardioprotection. Basic Res. Cardiol. 2011, 106, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Aletras, A.H.; Tilak, G.S.; Natanzon, A.; Hsu, L.Y.; Gonzalez, F.M.; Hoyt, R.F., Jr.; Arai, A.E. Retrospective determination of the area at risk for reperfused acute myocardial infarction with T2-weighted cardiac magnetic resonance imaging: Histopathological and displacement encoding with stimulated echoes (DENSE) functional validations. Circulation 2006, 113, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Varadharaj, S.; Ganesan, L.P.; Shobha, J.C.; Naidu, M.U.; Parinandi, N.L.; Tridandapani, S.; Kutala, V.K.; Kuppusamy, P. C-phycocyanin protects against ischemia-reperfusion injury of heart through involvement of p38 MAPK and ERK signaling. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2136–H2145. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.B.; Ahn, M.; Lee, S.M.; Koh, H.W.; Lee, S.H.; Kim, S.H.; Park, B.H. Inhibition of Janus activated kinase-3 protects against myocardial ischemia and reperfusion injury in mice. Exp. Mol. Med. 2013, 45, e23. [Google Scholar] [CrossRef] [Green Version]
- Luessi, F.; Kraus, S.; Trinschek, B.; Lerch, S.; Ploen, R.; Paterka, M.; Roberg, T.; Poisa-Beiro, L.; Klotz, L.; Wiendl, H.; et al. FTY720 (fingolimod) treatment tips the balance towards less immunogenic antigen-presenting cells in patients with multiple sclerosis. Mult. Scler. 2015, 21, 1811–1822. [Google Scholar] [CrossRef]
- Xu, H.; Jin, Y.; Ni, H.; Hu, S.; Zhang, Q. Sphingosine-1-phosphate receptor agonist, FTY720, restores coronary flow reserve in diabetic rats. Circ. J. 2014, 78, 2979–2986. [Google Scholar] [CrossRef] [Green Version]
- Markowski, P.; Boehm, O.; Goelz, L.; Haesner, A.L.; Ehrentraut, H.; Bauerfeld, K.; Tran, N.; Zacharowski, K.; Weisheit, C.; Langhoff, P.; et al. Pre-conditioning with synthetic CpG-oligonucleotides attenuates myocardial ischemia/reperfusion injury via IL-10 up-regulation. Basic Res. in Cardiol. 2013, 108, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonney, S.; Kominsky, D.; Brodsky, K.; Eltzschig, H.; Walker, L.; Eckle, T. Cardiac Per2 functions as novel link between fatty acid metabolism and myocardial inflammation during ischemia and reperfusion injury of the heart. PLoS ONE 2013, 8, e71493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Bian, Y.; Zhang, N.; Guo, J.; Wang, C.; Lau, W.B.; Xiao, C. Intermedin protects against myocardial ischemia-reperfusion injury in diabetic rats. Cardiovasc. Diabetol. 2013, 12, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Honbo, N.; Goetzl, E.J.; Chatterjee, K.; Karliner, J.S.; Gray, M.O. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3150–H3158. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013, 381, 166–175. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Heusch, G.; Musiolik, J.; Gedik, N.; Skyschally, A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ. Res. 2011, 109, 1302–1308. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline | Group A1 | Group B1 | p Value | Group A2 | Group B2 | p-Value |
|---|---|---|---|---|---|---|
| HR (beats/min) | 281 ± 16 | 278 ± 21 | Ns | 309 ± 29 | 295 ± 24 | ns |
| MAP (mmHg) | 137 ± 16 | 132 ± 18 | Ns | 112 ± 14 | 116 ± 16 | ns |
| CO (mL/min) | 42 ± 3.5 | 45 ± 4.2 | Ns | 44 ± 3 | 47 ± 5.2 | ns |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.; Laghari, A.H.; AlBkhoor, B.; Tabassum, S.; Meo, S.A.; Muhammad, N.; Linardi, D.; Al-Masri, A.A.; Fumagalli, G.; Luciani, G.B.; et al. Fingolimod Plays Role in Attenuation of Myocardial Injury Related to Experimental Model of Cardiac Arrest and Extracorporeal Life Support Resuscitation. Int. J. Mol. Sci. 2019, 20, 6237. https://doi.org/10.3390/ijms20246237
Ahmed N, Laghari AH, AlBkhoor B, Tabassum S, Meo SA, Muhammad N, Linardi D, Al-Masri AA, Fumagalli G, Luciani GB, et al. Fingolimod Plays Role in Attenuation of Myocardial Injury Related to Experimental Model of Cardiac Arrest and Extracorporeal Life Support Resuscitation. International Journal of Molecular Sciences. 2019; 20(24):6237. https://doi.org/10.3390/ijms20246237
Chicago/Turabian StyleAhmed, Naseer, Abid H. Laghari, Bashar AlBkhoor, Sobia Tabassum, Sultan Ayoub Meo, Nazeer Muhammad, Daniele Linardi, Abeer A. Al-Masri, Guido Fumagalli, Giovanni Battista Luciani, and et al. 2019. "Fingolimod Plays Role in Attenuation of Myocardial Injury Related to Experimental Model of Cardiac Arrest and Extracorporeal Life Support Resuscitation" International Journal of Molecular Sciences 20, no. 24: 6237. https://doi.org/10.3390/ijms20246237
APA StyleAhmed, N., Laghari, A. H., AlBkhoor, B., Tabassum, S., Meo, S. A., Muhammad, N., Linardi, D., Al-Masri, A. A., Fumagalli, G., Luciani, G. B., Faggian, G., & Rungatscher, A. (2019). Fingolimod Plays Role in Attenuation of Myocardial Injury Related to Experimental Model of Cardiac Arrest and Extracorporeal Life Support Resuscitation. International Journal of Molecular Sciences, 20(24), 6237. https://doi.org/10.3390/ijms20246237