1. Introduction
The unique physicochemical properties of engineered nanomaterials (ENMs) are being exploited for use in a growing variety of commercial nano-enabled products (NEPs), including electronics, cosmetics, agriculture and food, structural materials, as well as a wide variety of products for medical applications [
1,
2,
3,
4]. Numerous in vitro and in vivo studies have investigated the possible adverse effects of inhalation exposure to pristine ENMs during synthesis and handling [
5,
6,
7,
8]. However, human exposure is not limited to pristine ENMs, but to a wide variety of particles released from NEPs across their life cycle, including consumer use and disposal [
9,
10,
11,
12]. Indeed, the potential for exposure from such life cycle particulate matter (LCPM) may exceed that of pristine engineered nanoparticles (ENPs) [
13]. All pristine materials are ENPs. LCPM is the term we introduced to explain that pristine ENPs across their cycle and value chain can be released in the air but their properties differ due to their transformations. For instance, an ENP which will be used in a nano-enabled paint will be released during sanding and drilling, but the released LCPM will have different physico-chemical properties compared to pristine ENP used in the paint. The same applies to the pristine metallic nanoparticles used to formulate the toner of laser printers. When those ENPs are released in the air they will have completely different physico-chemical properties and will include in their surface organic compounds that are not there for the pristine ENPs. Moreover, the toxicological profiles of LCPM may differ greatly from those of pristine ENPs [
14,
15]. Despite the potential for LCPM exposure, most nanotoxicology studies have focused on pristine ENPs and thus, toxicological evaluation of LCPM has been limited. The National Institute for Occupational Safety and Health (NIOSH) and the Environmental Protection Agency (EPA) have, therefore, recommended life cycle analyses in their nanotechnology research programs [
16,
17]. Most importantly, we need to develop integrated methodologies that can link real world LCPM exposures to toxicology and diseases.
The rate at which production of new ENPs and incorporation into NEPs occurred has far exceeded our ability to test all ENPs and LCPM released over the NEP life cycle using in vivo animal studies. This study will focus on a specific real-world LCPM: laser printer-emitted particles (PEPs) generated from nano-enabled toners which has been studied extensively by the authors and others over the last decade [
18,
19]. In US alone, 23 million printers are produced annually, and over 160,000 workers are employed in copier centers [
20,
21,
22,
23,
24,
25,
26,
27,
28]. The new generation of toners contain ENPs, such as metals and metal oxides, and that PEPs with mode sizes of 49–208 nm are released during printing at concentrations of up to 1.3 million particles/cm
3 [
15]. There is growing evidence from both in vitro cellular and in vivo animal studies of significant bio-activity of PEPs, which points to potential adverse health effects from inhalation exposure [
15,
28,
29,
30,
31,
32,
33,
34,
35,
36]. Our group recently suggested exposure to PEPs increases cardiovascular risk by impairing ventricular performance and inducing hypertension and arrhythmia [
37]. Currently, there are no available biomarkers for assessing adverse cardiovascular responses induced by PEPs exposure in susceptible individuals with long-term exposure.
Genomic/transcriptomic analysis has become a routine approach in biomedical research and has increasing applications in toxicology [
38,
39]. However, this is not the case in nanotoxicology research. Gene expression analysis could aid mechanistic studies by evaluating the relevance of signaling pathways representing significantly perturbed genes for treatment interventions [
40,
41,
42]. Since miRNAs are more stable than mRNA, and can be obtained from minimally-invasive biological samples, such as blood, they can provide more insight into identifying valuable diagnostic and predictive biomarkers [
43,
44]. Integrated analysis of miRNA and mRNA could reveal post-transcriptional functional involvements of the identified gene markers and provide novel biomarkers for worker surveillance and targets for therapeutic interventions of cardiovascular disease or lung damage [
45,
46]. Schulte et al. [
47] suggested that biomarkers of early responses are necessary for an optimal risk surveillance program for workers in the nanotechnology field.
This study will address this research gap using a systematic, integrated approach combining in vivo, metabolomic, lipidomic, and transcriptomic studies, to evaluate both pulmonary and cardiovascular effects of PEPs exposure. Metabolomic profiles were sequentially assessed following exposure to PEPs to reflect dynamic responses to any pathophysiological changes that may have occurred. Such an approach provides a fresh opportunity for biomarker discovery to elucidate systemic changes occurring following exposure to PEPs. Genome-scale profiles were performed in rat lung tissue and blood to identify important gene signatures relevant to PEPs-induced adverse effects and potential mechanistic pathways. The highly novel integrated in vivo and computational transcriptomic/metabolomic analyses in this study will identify biomarkers of potential PEPs-induced adverse cardiopulmonary effects for occupational/commercial risk surveillance, and reveal important insights into intervention strategies.
3. Discussion
PEPs generated from nano-enabled toners are one of the most common types of LCPM released from various nano-enabled products in consumer and occupational use scenarios. The toxicological assessment of PEPs is of utmost importance for worker and consumer health protection. PEPs are deemed highly bioactive in in-vitro and in-vivo studies and may impose significant potential adverse health effects, as previously reported by our group and others [
15,
28,
29,
30,
31,
32,
33,
34,
35,
36]. This study utilized a highly novel and integrated, genome-wide mRNA and miRNA profiling in rat lung and blood combined with metabolomics and lipidomics profiling in rat serum to assess PEPs-induced cardiopulmonary risk and other disease implications. The identified PEPs-induced dysregulated genes, pathways, biological processes, molecular functions, and miRNA-mediated transcriptional activities provide important insights into the disease mechanisms and are in agreement with in-vitro and in-vivo toxicological studies of PEPs. Furthermore, the identified important mRNA/miRNA, lipids and metabolites may serve as candidate biomarkers for future occupational and medical surveillance studies.
The respiratory system is the primary route of exposure to airborne particulate matters (PM) via inhalation. Epidemiological studies indicated there is a correlation between inhalation of particles from ambient air and an increased incidence of cardiovascular diseases [
50,
51]. Numerous in vivo and in vitro studies demonstrated that inhalation exposure to PM causes not only pulmonary damages, but induces adverse cardiovascular effects [
52,
53,
54]. Several mechanisms may be involved in the induction of cardiovascular effects upon inhalation exposure of particulates, particularly those less than PM0.1. First, the translocation of particulates from respiratory system into the blood circulatory through penetrating the air-blood barrier [
55]. Second, inhalation of particulates induces pulmonary inflammation, which may mediate a systemic oxidative/inflammatory response that leads to cardiovascular effects [
56]. Third, inhalation exposure may trigger neurogenic regulated mechanisms to modulate cardiovascular function [
57]. Therefore, evaluation of the changes in gene expression of lung and blood following inhalation exposure to particulates in printer emissions may provide a transcriptomic profiling tool to assess cardiovascular toxicity as well as potential toxicity in other tissue organs. The findings from this study will provide guidance for designing future specific disease model-based studies of PEPs-induced toxicity in a particular tissue organ.
Our group reported that PEPs exposure caused no damage in the histological and chemiluminescence analysis of lung and heart tissues in the in vivo animal studies [
48]. However, PEPs exposure elevated sympathetic influence, impaired ventricular performance and repolarization, and caused hypertension and arrhythmia, suggesting increased cardiovascular risk [
37]. The results of the global transcriptomic profiling in this study confirmed these pathological data. No significant perturbation in molecular pathways or processes linking to pulmonary diseases was identified in rat lung or blood following PEPs inhalation. In contrast, PEPs inhalation exposure dysregulated genes and pathways involved in cardiovascular malfunction at every observed time point in rat lung and several time points in rat blood. At day 1 of PEPs exposure,
Fgb was up-regulated (
p < 0.05) in rat lung, with implications in inherited thrombophilia, afibrinogenemia and dysfibrinogenemia (
Table S2). At day 5 of PEPs inhalation,
RT1-Bb, associated with dilated cardiomyopathy, viral myocarditis, and Type I diabetes mellitus, was down-regulated (
p < 0.05) in rat lung (
Table 1 and
Table S2). At day 9 of PEPs exposure,
Scn5a was up-regulated (
p < 0.05) in rat lung, indicating implications in familial idiopathic ventricular fibrillation, dilated cardiomyopathy, progressive cardiac conduction defect, and progressive familial heart block (
Table S2). At day 13 of PEPs inhalation, genes involved in the regulation of vascular endothelial growth factor production,
Noda1,
Sulf2, and
Ccr2, were down-regulated (
p < 0.05) in rat blood (
Table S2). At day 21 of PEPs inhalation,
Tnnc1 was down-regulated (
p < 0.05) in rat lung, with involvements in cardiac muscle contraction and tissue morphogenesis, dilated cardiomyopathy, and hypertrophic cardiomyopathy (
Table 1 and
Table S2).
Myh6, associated with left ventricular noncompaction, was down-regulated (
p < 0.05) in rat lung (
Table S2). These results are concordant with the cardiovascular pathology data (manuscript in review [
37]).
PEPs inhalation exposure also dysregulated ATP activities, sodium and manganese metabolism, and redox functions in rat lung and blood with implications in cardiovascular dysfunctions and blood pressure abnormality. In PEPs exposed rat blood, up-regulation of
Atp6v0e1 at days 1 and 13, down-regulation of
Atp5g1 and
Atp6v1f at day 1, and up-regulation of
Atp6v1g3 at day 13 were evident (
p < 0.05;
Table S3). Five days of continuous PEPs inhalation up-regulated
Slco3a1 and
LOC685081 and down-regulated
Slco5a1, which are associated with sodium-independent organic anion transmembrane transporter activity (
p < 0.05;
Table S3). Perturbed aldosterone-regulated sodium reabsorption with up-regulation of
Kras was identified in rat blood at day 9 of PEPs inhalation (
p < 0.05;
Table 3). Manganese ion binding dysfunction with up-regulation of
Ppef2 and down-regulation of
Idi1 was identified in rat lung at day 9 of PEPs exposure (
p < 0.05;
Table S2). These molecular dysfunctions of sodium and manganese absorption are relevant to high blood pressure, and confirm the observed blood pressure variation in PEPs-exposed rats (manuscript in review [
37]). Remarkable disrupted redox functions were identified in rat lung and blood following PEPs inhalation. At day 1, altered oxidoreductase activity and arachidonic acid monooxygenase activity with up-regulation of
Cyp2c22,
Cyp3a18,
Cyp2b3,
Cyp2a1,
Cyp2c6v1,
Cyp4a3 and
Cyp3a23/3a1 were identified in rat lung (
p < 0.05;
Table S2). At 17 days of continuous PEPs inhalation, negative regulation of oxidoreductase activity was ranked a top changed biological process in rat blood with up-regulation of
Cav1 and
Hp (
p < 0.05;
Table S3).
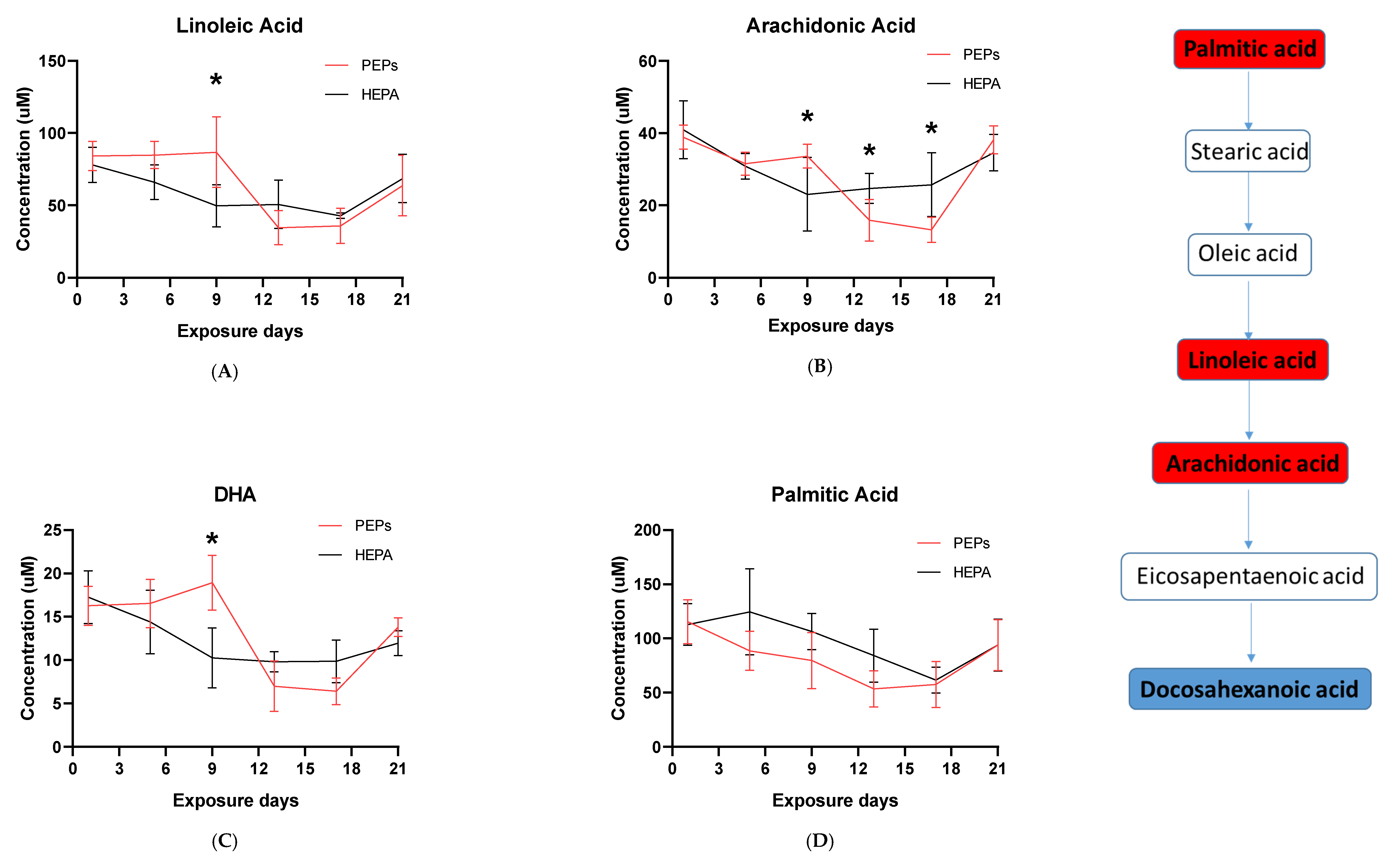
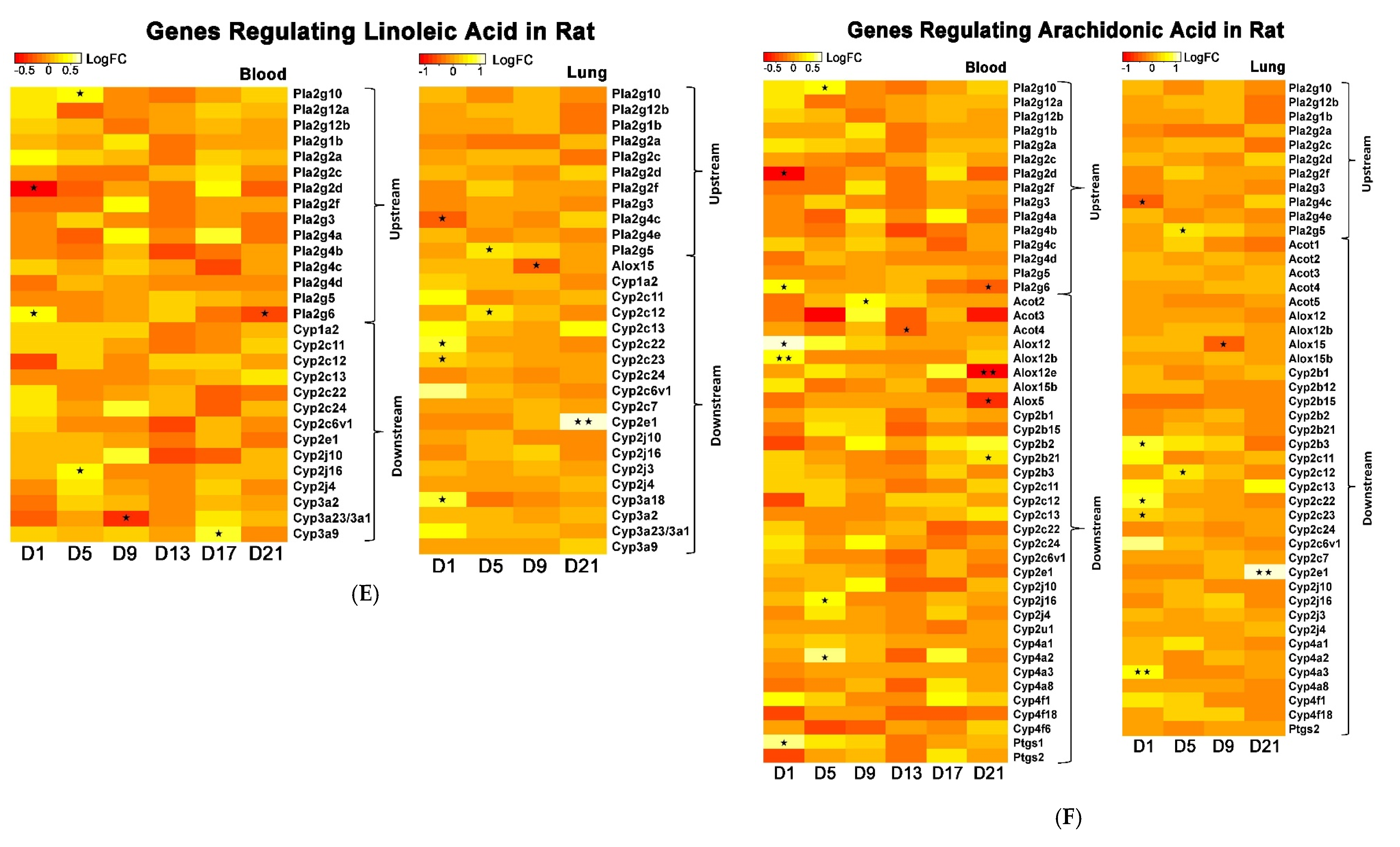
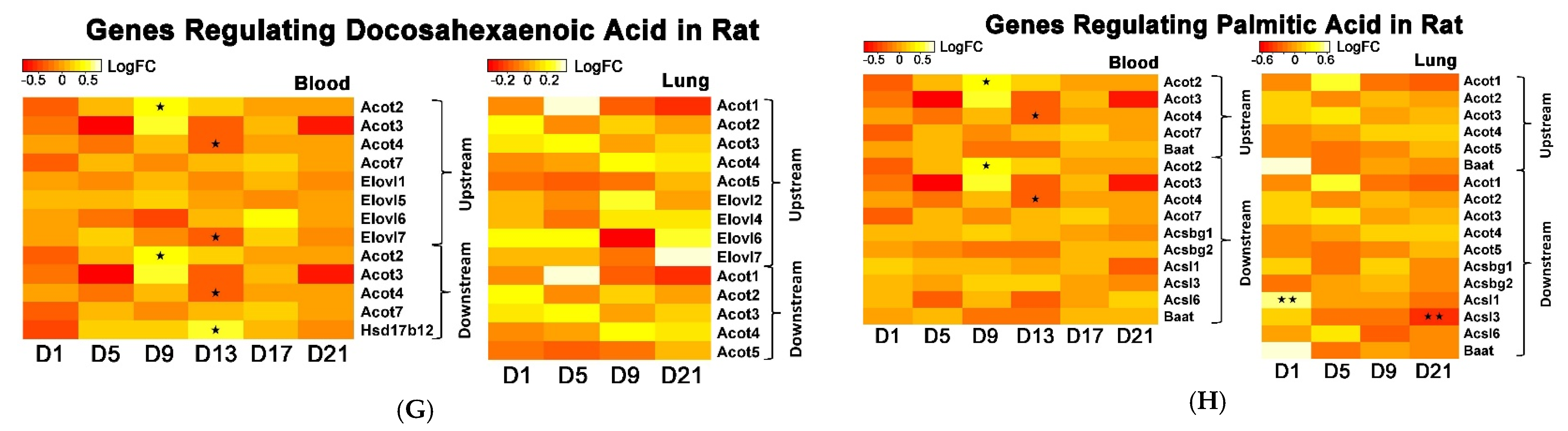
The whole genome analysis mirrored the global metabolomics profiles following whole-body PEPs inhalation. Global metabolomics illustrated significant (
p < 0.05) activation of glycerophospholipid metabolism, linoleic acid metabolism, and biosynthesis of unsaturated fatty acids upon PEPs exposure. In transcriptomics analysis, linoleic acid metabolism pathway was among top 5 significant (
p < 0.05) pathways in rat lung at Day 1 of PEPs inhalation, with up-regulated genes
Cyp2c6v1,
Cyp2c22,
and Cyp3a18 (
p < 0.05;
Table 1). The up-regulation of these 3 genes was also involved in dysregulated steroid hormone biosynthesis, epoxygenase P450 pathway, and endopeptidase activity (
Table S2). At Day 5 of PEPs inhalation, biosynthesis of unsaturated fatty acid pathway was a top altered pathway in rat blood, with up-regulation of
Hacd1 and down-regulation of
Acaa1a (
p < 0.05;
Table 3). In addition, negative regulation of fatty acid metabolic process was evident in rat blood at Day 17, with up-regulation of
Insig2 (
p < 0.05;
Table S3).
Activation of the innate immune systems via extracellular and/or intracellular receptors, such as Fc, mannose or Toll-like receptors, can lead to the synthesis of inflammatory lipid mediators like eicosanoids [
58,
59]. This is in agreement with in-vitro PEPs studies by the authors [
60]. These lipid mediators are derived from arachidonic acid, an ω-6 polyunsaturated fatty acid that mammals directly incorporate through diet or by synthesis from linoleic acid [
61]. Prostanoids, a major class of eicosanoids, modulates the inflammatory and immune responses [
59]. Prostaglandin (PG) E2 is by far the best characterized prostanoid from an immunomodulatoty perspective. Although cytokines are key regulators of immunity, eicosanoids, including PGE2, PGD2, leukotriene (LT) B4, including LTC4, also differentially affect the immune cells by modulating cytokine release, cell differentiation, survival, migration, antigen presentation, and apoptosis. By acting on various aspects of immune function and inflammation, such lipid mediators act as key regulators of the crosstalk between innate and adaptive immunity. Upon early exposure to PEPs (Days 1–9), an up-regulation in linoleic acid, arachidonic acid and DHA metabolism was detected, suggestive of an innate immunity response to PEPs. Particle-generated reactive oxygen species (ROS) can further influence signaling pathways leading to activation of the arachidonic acid cascade [
62,
63,
64]. Studies of titanium oxide nanoparticle inhalation shows significantly impairment of the endothelium-dependent vasoreactivity in coronary arterioles due to increases in microvascular ROS [
65]. Similarly, in a previous in-vitro PEPs study by the authors, it was shown that small airway epithelial cells exposure induces cellular effects on human microvascular endothelial cells in an alveolar-capillary co-culture model [
28]. The addition of prostanoid formation further contributed to this dysfunction. Deregulation in coronary microvascular function may contribute to cardiac events associated with exposure to particles in this size range [
65]. Similar reports have also describe the impact on the arachidonic acid pathway by combustion-derived particles and air pollution [
62,
66].
Arachidonic acid may also be processed through non-enzymatic reactions. The four double bonds of arachidonic acid are readily oxygenated to form bioactive molecules. Therefore, oxidative stress and/or exposure to ROS and reactive nitrogen species (RNS) results in the oxidation of arachidonic acid, leading to generation of isoprostanes and nitroeicosatetraenoic acids [
67], which in turn inhibits COX1, and causes platelet aggregation, vasoconstriction, smooth muscle cell proliferation, and cardiomyocyte hypertrophy [
67]. Arachidonic acid enters numerous metabolic pathways that interconnect lipid metabolism with immunity and, therefore, has widespread effect.
DHA possesses anti-inflammatory functions [
68]. Resolvins, whose functional roles involve promoting restoration of normal cellular function following inflammation after tissue damage, are a metabolic by-product of omega-3 fatty acid [
69,
70]. High DHA levels associate with linoleic and arachidonic acid, suggesting an association with a resolving inflammatory response following PEPs inhalation. Inflammation peaked at Day 9 and was suppressed from Day 13 to Day 17 followed by high DHA expression which returned to baseline by Day 21 (
Figure 2C).
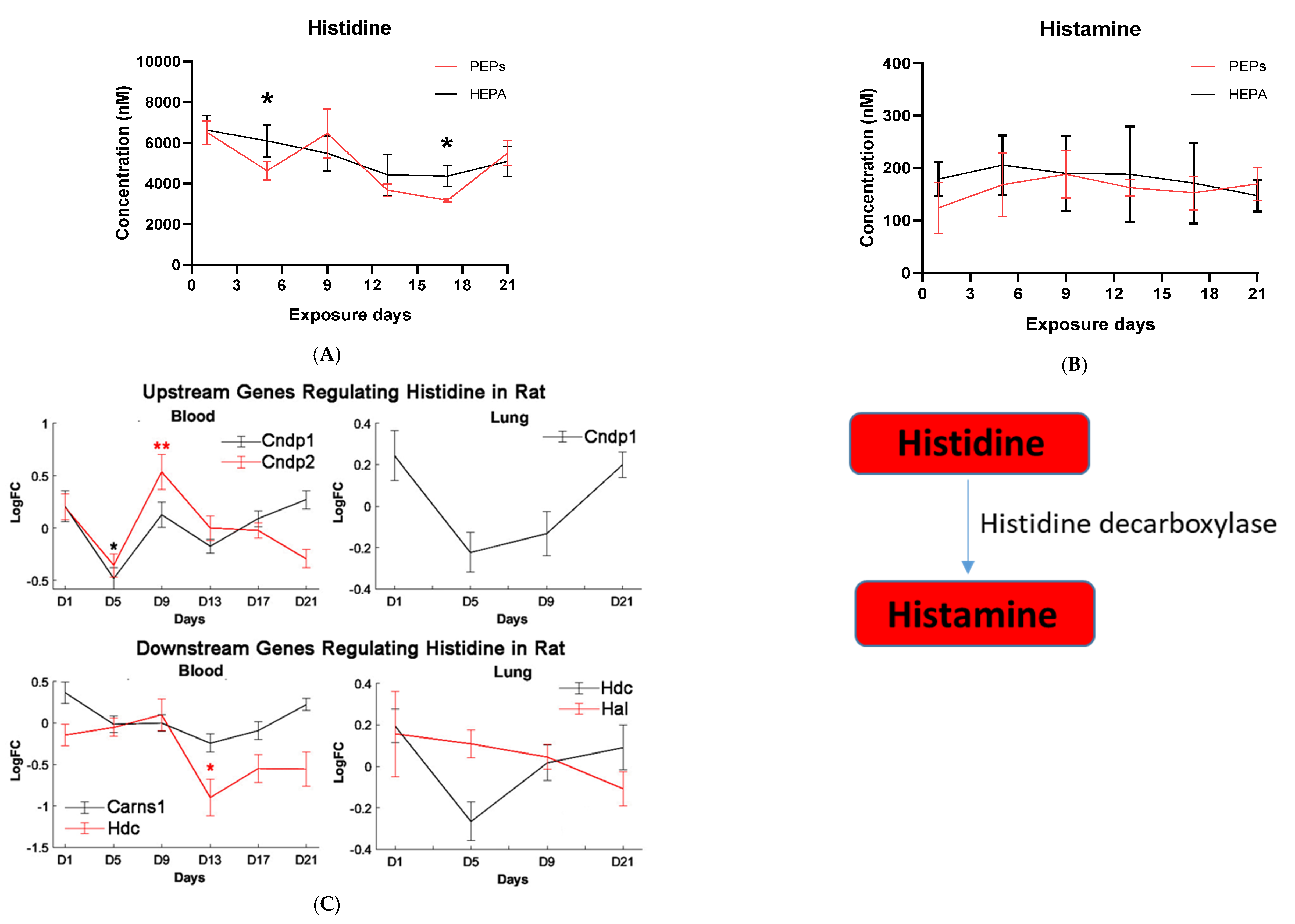
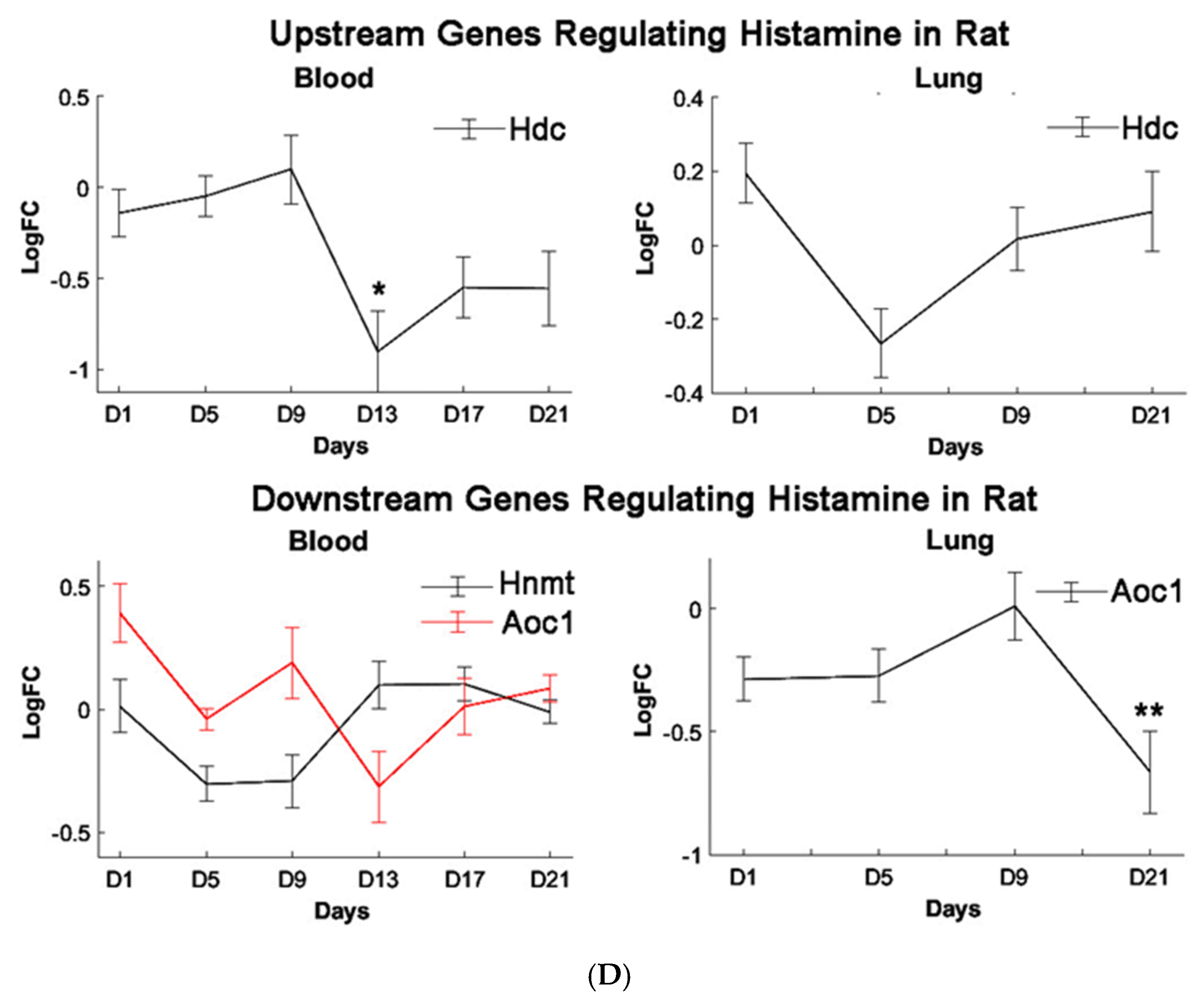
Histidine is the precursor of histamine production. Metabolomic change in both is, therefore, of interest due to their role in allergen-related sensitization and inflammation [
71]. Despite this, a significant decrease in histidine levels, but not histamine levels, was observed in our metabolomic profiles, suggesting PEPs may have minimal roles in inciting the allergic response. Prior studies have shown that L-histidine exhibits antioxidant capabilities, such as scavenging free radicals and chelating divalent metal ions, hence its use to manage oxidative stress [
72,
73,
74]. The physiological effects of histidine include inhibition of pro-inflammatory biomarkers [
74]. In vivo and in vitro studies illustrate that administration of histidine lowers the levels of pro-inflammatory markers, such as interleukin 8, interleukin 6, C-reactive protein and tumor necrosis factor alpha [
75,
76,
77]. This indicates that histidine does more than merely scavenge ROS but directly influences pro-inflammatory cytokines [
75,
76,
77]. Mechanisms are likely associated with its potential to increase the amino acid pool and the antioxidant status of cells [
78]. Histidine levels are low in patients with heart failure and are correlated with B-type natriuretic peptide, left ventricle ejection fraction, left ventricle end-diastolic volume index, inferior vena cava diameter and the ratio of early diastolic velocity of the mitral inflow to mitral annulus, respectively [
79]. L-histidine is an effective inhibitor of spontaneous platelet aggregation and platelet TXB2 generation and
l-histidine intake reduces spontaneous platelet aggregation [
80]. Such change likely contributes to ROS attenuation in tissues and alleviate clinical symptoms while protecting against intestinal, nervous, and cardiovascular damage.
4. Materials and Methods
4.1. Whole-Body Inhalation Exposure of Animals
The details of animal exposure and PM characterization were described by the authors in a recent study [
48]. In summary,
Figure S1 (adapted from Pirela et al. [
48]) outlines the Printer Exposure Generation System (PEGS) used for the animal exposure. The PEPs-exposed group consisted of Sprague Dawley rats housed in individual cages and exposed via whole-body inhalation to the PEPs and gaseous pollutants of laser printer B1 using the PEGS described in detail in a previous publication [
48]. The printer was used to generate PEPs by printing a 5%-page coverage monochrome document using standardized settings [
15]. The laser printer B1 was selected based on exposure and toxicological profiles reported in our previous studies [
15]. In parallel, another group of animals was exposed to high-efficiency particulate air (HEPA)-filtered air. The system was configured for twelve chambers (one animal per chamber), each equipped with a port for aerosol delivery and temperature and humidity measurement, and a gas sampling port.
Animals were placed in their chambers for 1–5 h for 5 days prior to the exposures to ensure they gradually acclimated to the exposure chambers. The following week, animals were exposed to either PEPs or the room HEPA-filtered air 5 h/day for up to 21 consecutive days. The exposure duration of 21 days, as well as the sacrifice time points (Day 1, 5, 9, 13, 17, and 21), were chosen as sub-chronic exposure periods to provide several delivered doses to the lung and identify any signs of toxicity in the cardiovascular or respiratory systems.
4.2. Animal Exposure Characterization (PEPs and Gaseous Co-Pollutants)
Real-Time Measurements
The particle number concentration, size distribution, temperature, relative humidity and total volatile organic compounds (tVOC) levels were measured in real time in one of the 12 animal inhalation exposure chambers throughout the exposure durations. A scanning mobility particle sizer (SMPS Model 3080, TSI Inc., Shoreview, MN, USA) was also used for measuring particle number concentration and size distribution (ranging from 2.5 to 210 nm) in the chamber. Real-time particle number concentration as a function of size collected by the SMPS was used to estimate the mass size distribution assuming spherical particles and the density of carbon. Real-time tVOC levels were also monitored using a tVOC monitor (Graywolf Sensing Solutions, Shelton, CT, USA). All the realtime instruments were calibrated, and background tests were performed at the beginning of each sampling experiment. No significant variation in the temperature (°C) and relative humidity (%) in the inhalation animal chambers was observed throughout the exposure period [
48].
It is worth noting that a comprehensive chemical analysis of PEPs was performed by the authors and described in detail in previous publications [
19,
21,
48]. In summary, PEPs possess a complex mixture of metals (1–33%) and primarily various organic compounds including polycyclic aromatic hydrocarbon (PAHs; 0.0067%) and other compounds such as elemental carbon (1–3%), organic carbon (42–89%). It is also worth noting that in a recent publication by the authors [
19], it was documented that there are synergistic interactions of catalytic metallic nanoparticles with gaseous sVOCs co-pollutants which result in the formation of high molecular and carcinogenic PAHs on the surface of the PEPs.
4.3. Animals
One hundred thirty-four healthy, male, 9-week-old Sprague-Dawley rats, weighing an average of 300 g, were purchased from Taconic Farms Inc. (Hudson, NY, USA). The rats were allowed to acclimatize for 1 week before the studies were initiated. Baseline data was collected for 1 week before the exposures occurred. The rats were maintained on a 12-h light/dark cycle and food and water were provided ad libitum. Upon arrival, animals were randomly assigned a unique identification number, which determines the exposure group for the animal. There were two treatment groups: (a) Control group: high-efficiency particulate air (HEPA) filtered air, and (b) PEPs exposure group. All the animals were treated humanely in accordance with the guiding principles on the Use of Animals in Toxicology and the Animal Care and Use Committee of Harvard University (Ethical permit No. 04623, 2015).
4.4. Whole Blood and Lung Tissue Collection
At the completion of the exposure periods, the animals in each group were anaesthetized with an intraperitoneal injection of a lethal dose (200 mg/kg) of pentobarbital sodium (Anthony Products Co., Arcadia, CA, USA) and sacrificed by exsanguination, followed by whole blood and lung tissue collection. Blood was collected in microfuge tubes pre-loaded with RNAlater and inverted 10 times and transferred to −20 °C or −80 °C for storage. The samples were extracted according to the manufacturer’s instructions or transferred to −80 °C for long term storage. The lung tissue was removed, placed in liquid nitrogen, and stored at −80 °C.
4.5. RNA Isolation
Total RNA from frozen lung tissue was extracted using the RNeasy kit, with simultaneous on-column digestion of DNA during RNA purification according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Total RNA isolation from whole blood (stored in RNAlater) was carried out using RiboPure-Blood kit, followed by DNA removal treatment as described by the manufacturer (LifeTechnologies, Carlsbad, CA, USA). Concentration and purity of RNA were quantified by the A 260 nm/A 280 nm and A 260 nm/A 230 nm ratios using a NanoDrop ND-100 spectrophotometer (Thermo Scientific lnc., Waltham, MA, USA). Samples were stored at −80 °C until shipment to the University of Michigan for further processing and analysis.
4.6. mRNA and miRNA Microarray Profiling
Global mRNA expression profiles were generated with Rat Gene ST 2.1 plates at the University of Michigan Microarray Facility using an Affymetrix Plus kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Global miRNA expression profiles were generated with Rat GeneChip miRNA4.1 plates with the same total RNA samples using a FlashTag kit. cDNA was synthesized from 500 ng total RNA, amplified, fragmented, and biotinylated using a GeneChip WT PLUS Reagent kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to manufacturer’s instructions. cDNA was then prepared for hybridization with reagents from the Affymetrix GeneTitan Hybridization, Wash, and Stain Kit for WT Array Plates. For hybridization, 2.76 µg was hybridized to the Affymetrix Rat Gene ST 2.1 Arrays, which were then washed, stained, and scanned using the GeneTitan Multi-Channel Instrument according to Affymetrix’s User Guide for Expression Array Plates (P/N 702933 Rev. 2, 2013). Data were analyzed using the Limma, Oligo, and Affy Bioconductor packages implemented in the open source R statistical environment. The robust multi-array average was used to normalize the data and fit log2-transformed expression values. The raw microarray data were processed using a robust multi-array average (RMA) method, and expression values were log2 transformed, with a principal component analysis used as the final quality control step to visualize mRNA expression values. The analysis was performed with the oligo package of Bioconductor in the R statistical environment. The mRNA and miRNA expression profiles in rat lung and blood are available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) with accession number GSE127951 (with Sub-Series GSE127945, GSE127947, GSE127949, and GSE127950).
4.7. mRNA and miRNA Data Analysis
Affymetrix
Transcriptome Analysis Console (TAC) software was used in miRNA and mRNA data analysis. The microarray data were analyzed with weighted linear models and Bayes methods designed specifically for microarray analysis [
81]. Samples were also weighted based on a gene-by-gene update algorithm designed to downweight chips that are deemed less reproducible [
82]. Probe sets that had a variance less than 0.05 were removed from further analysis. For mRNA data, only the probe sets listed as “main” by Affymetrix were included for further pathway analysis, which excluded internal controls and un-annotated genes. Probe sets with a fold change of 1.5 or greater were selected.
p-Values were adjusted for multiple comparisons using the false discovery rate (FDR) with Benjamini and Hochberg tests.
4.8. Pathway, Gene Ontology, and Disease Analysis
Significant differentially expressed genes in PEPs exposed vs. HEPA-filtered air exposed rat lung and blood were analyzed with iPathwayGuide (Adviata; Plymouth, MI, USA). These genes were obtained using a threshold of 1 for statistical significance (
p-value) and a log fold change of expression with the absolute value of at least 0.58. The significance is represented in terms of the negative log (base 10) of the
p-value. These data were analyzed in the context of pathways obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Release 84.0 + /10–26 October 17) [
83], gene ontologies (GO) from the Gene Ontology Consortium database (6 November 2017) [
84], miRNAs from the miRBase (Release 21) and MICROCOSM (MicroCosm Targets Version 5) databases [
85], and diseases from the KEGG database (Release 84.0 + /10–26 October 17) [
83]. The top 5 pathways were ranked in terms of the two types of evidence computed by iPathwayGuide: over-representation on the x-axis (pORA) and the total pathway accumulation on the y-axis (pAcc). The two types of evidence, pORA and pAcc, were combined into one final pathway score by calculating a
p-value using Fisher’s method. This
p-value was then corrected for multiple comparisons using FDR or Bonferroni corrections.
4.9. Prediction of Active miRNAs
The prediction of active miRNAs [
86] is based on the enrichment of differentially downregulated target genes of the miRNAs. iPathwayGuide calculates the probability of observing a greater number of differentially downregulated target genes for a given miRNA just by chance. This
p-value was computed using the hypergeometric distribution.
4.10. Lipid and Metabolite Profiling of Rat Serum
4.10.1. Sample Processing
Plasma (100 µL) and 400 µL of lipid/internal standard in isopropanol or acetonitrile were mixed and incubated at 4 °C, with constant agitation at 1400 rpm for 2 h following centrifugation at 4 °C at 4500 rpm for 10 min. Supernatants were then collected for analysis [
87].
4.10.2. Metabolite Quantification
One hundred microliters of serum were mixed with 200 µL of methanol-water (50:50 v/v) by vortexing for 30 s. Following this, the mixture was incubated at 4 °C for 1 h and added to 200 µL chloroform. Following vigorously vortexing (30 s) and centrifugation (14,000 rpm for 10 min at 4 °C), proteins were precipitated. The organic and aqueous phases were next collected separately and dried using vacuum centrifugation. The aqueous phase was then reconstituted in 200 µL of mobile phase E:F (25:75 v/v, mobile phase E-30 mM ammonium formate in water and mobile phase F-30 mM ammonium formate in 85:15 ACN: Water) and used for histamine and histidine analysis. The organic phase was reconstituted in 100 µL ethanol for fatty acid analysis.
4.10.3. Histamine and Histidine Analysis
LC-MS/MS histamine and histidine analysis was performed using an ACQUITY UPLC system (Waters, Milford, MA, USA) coupled to a Xevo TQ-MS system (Waters, Milford, MA, USA). Sample (10 µL) were separated using A Waters ACQUITY BEH Amide (1.7 µM; 2.1 × 150 mM) at 40 °C in ESI Positive mode for the aqueous phase containing histamine and histidine with eleution gradient using mobile phases C and D (flow rate 0.4 mL/min) at 75% D linearly decreasing to 45% for 2 min and held for 1 min before equilibrating the system for the next injection. For mass spectrometry, we used a capillary voltage of 3.4 kV, desolvation temperature 400 °C, cone gas flow 150 L/h, desolvation gas flow 800 L/h and a source temperature of 150 °C for analysis. Corresponding parameters for histamine and histidine analysis were 2.5 kV, 450 °C, 0 L/h, 900 L/h, and 150 °C, respectively. MassLynx and TargetLynx were used for data acquisition quantification respectively. Statistical analysis was carried out using 2-way ANOVA followed by post-hoc multiple correction using FDR [
88]. Details of lipid and metabolite profiling were included in
Supplementary File.

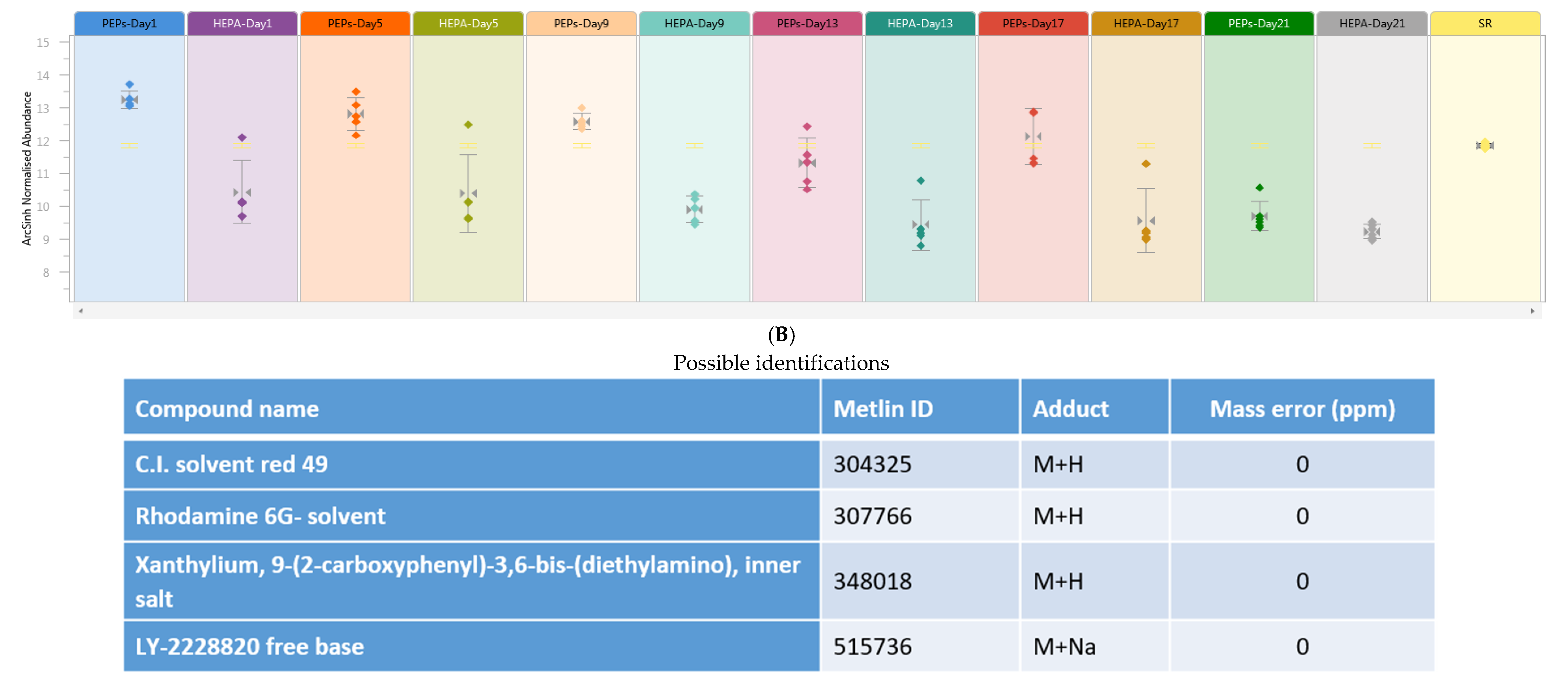
 ,
, 








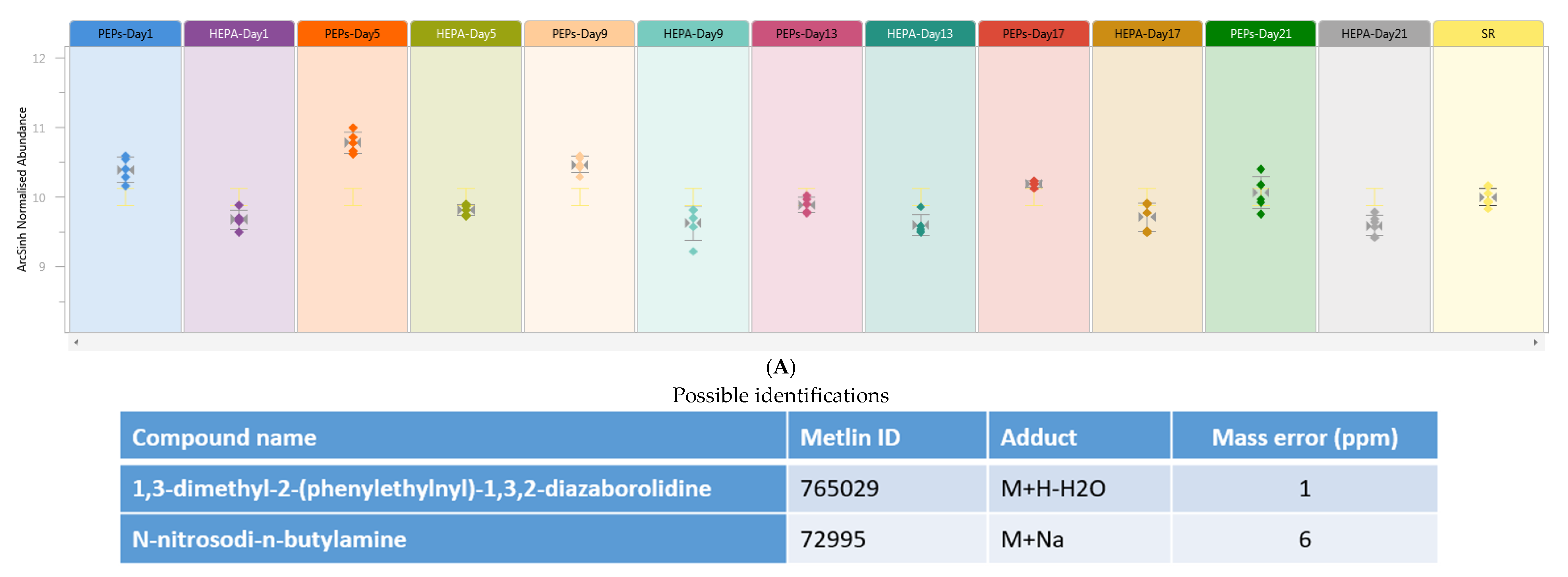{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}