Zn2+ Aggravates Tau Aggregation and Neurotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
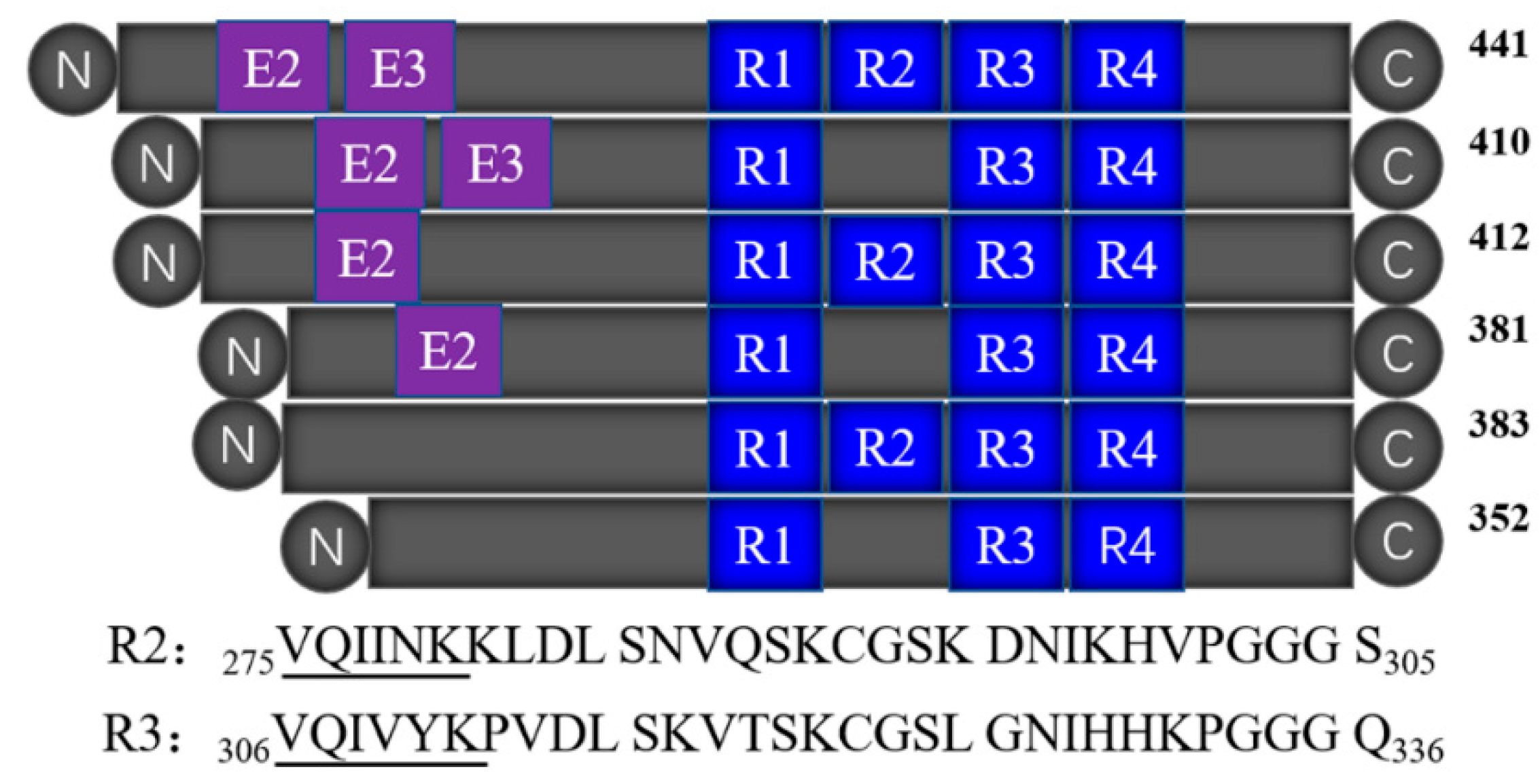
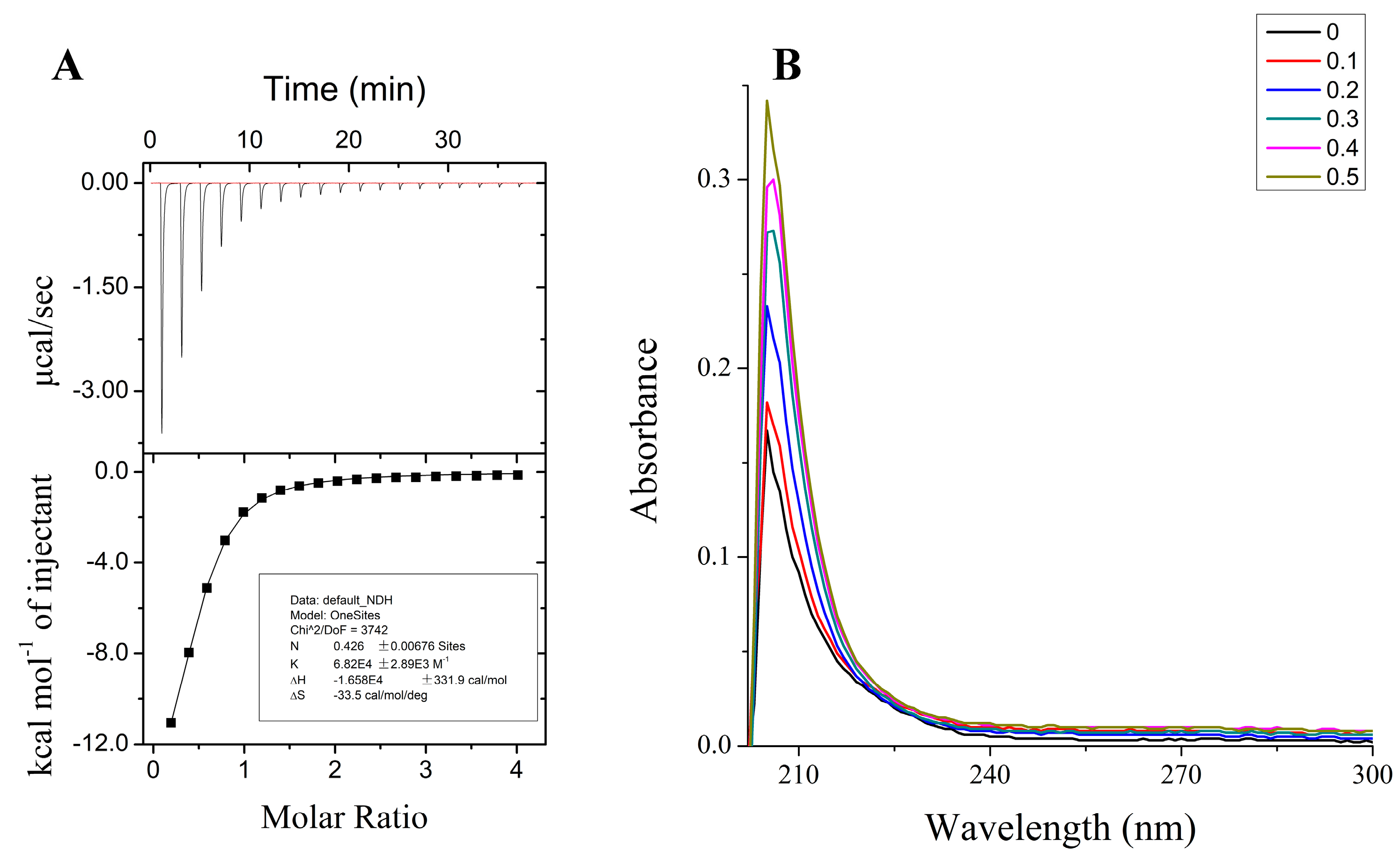
2.1. Tau-R3 Bound Zn2+ with Moderate Affinity
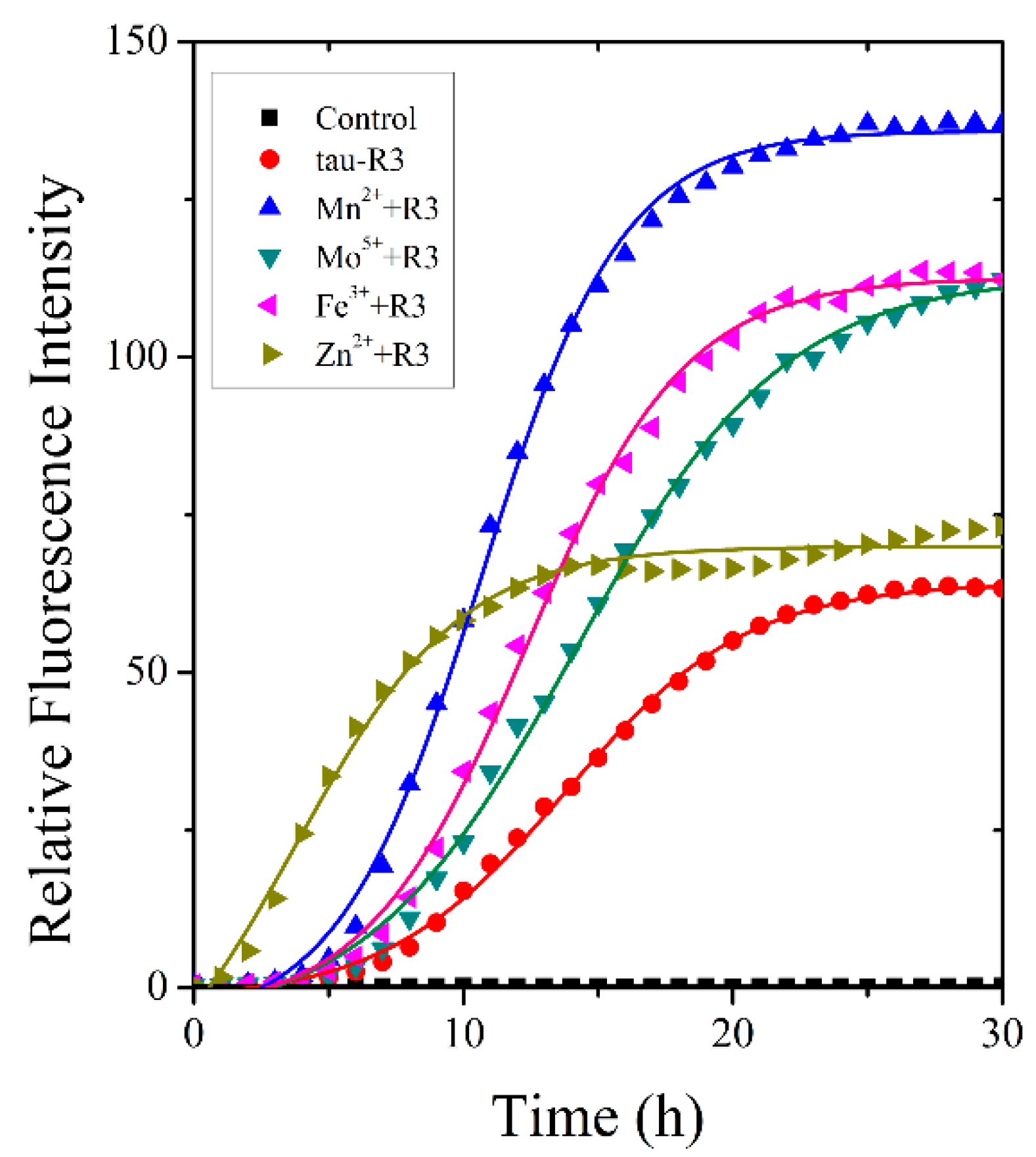
2.2. Zn2+ Accelerated the Fibrillization of Tau-R3 In Vitro
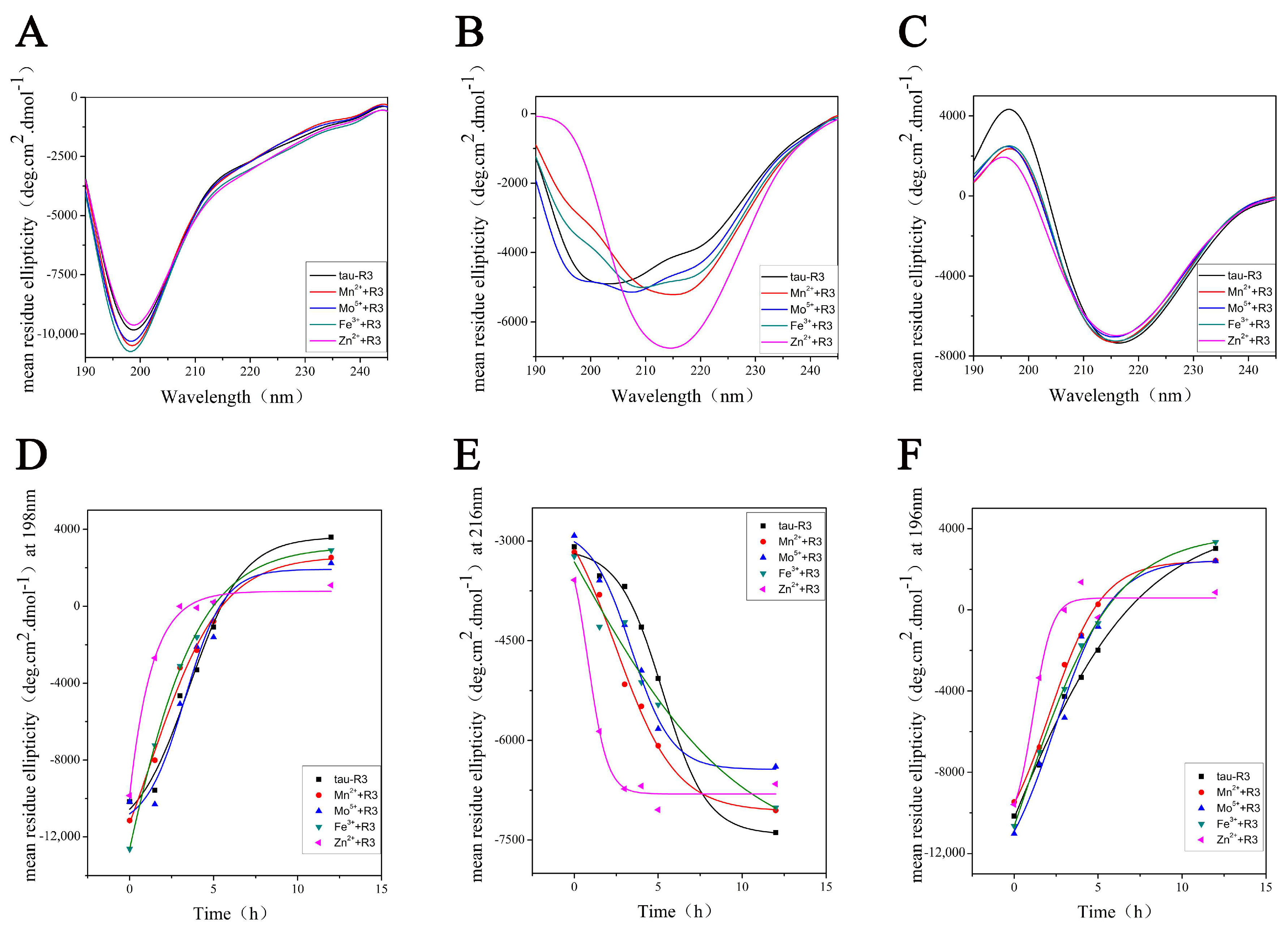
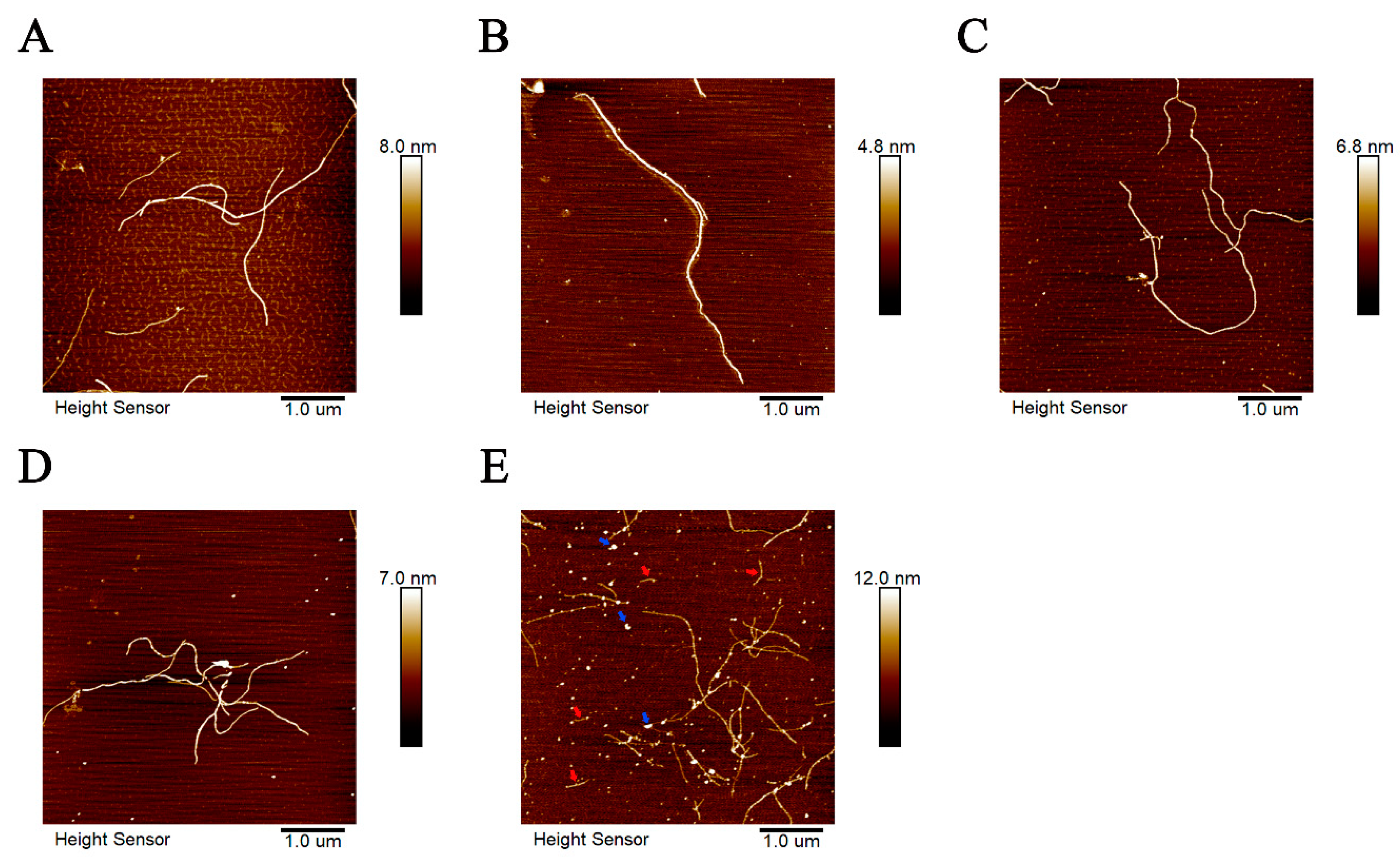
2.3. Zn2+ Changed the Morphology of Tau Aggregates
2.4. Zn2+ Aggravates Tau-R3-Mediated Toxicity and ROS Induction in Nerve Cells
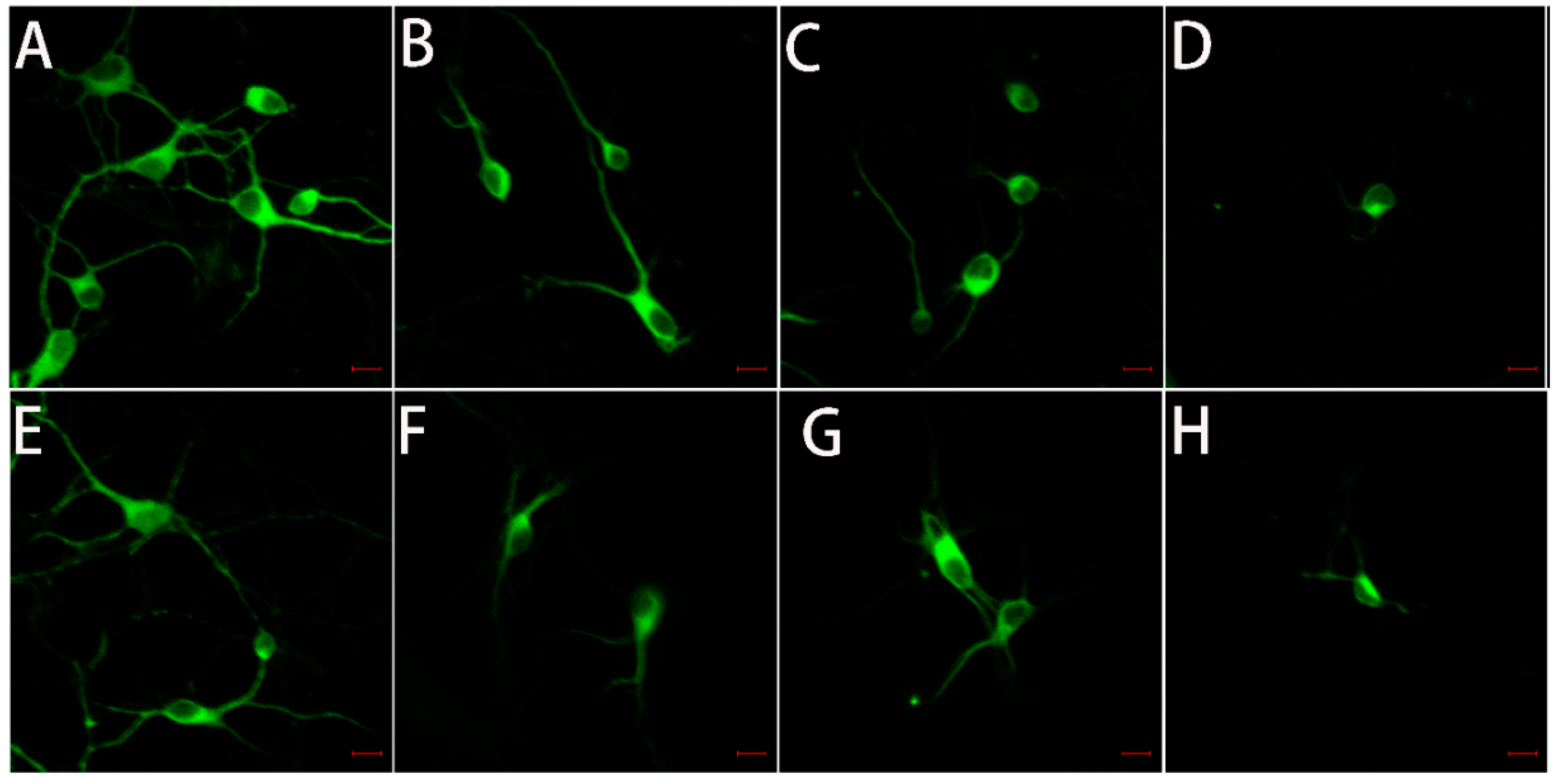
2.5. The Toxicity of Tau-R3 to Primary Neurons
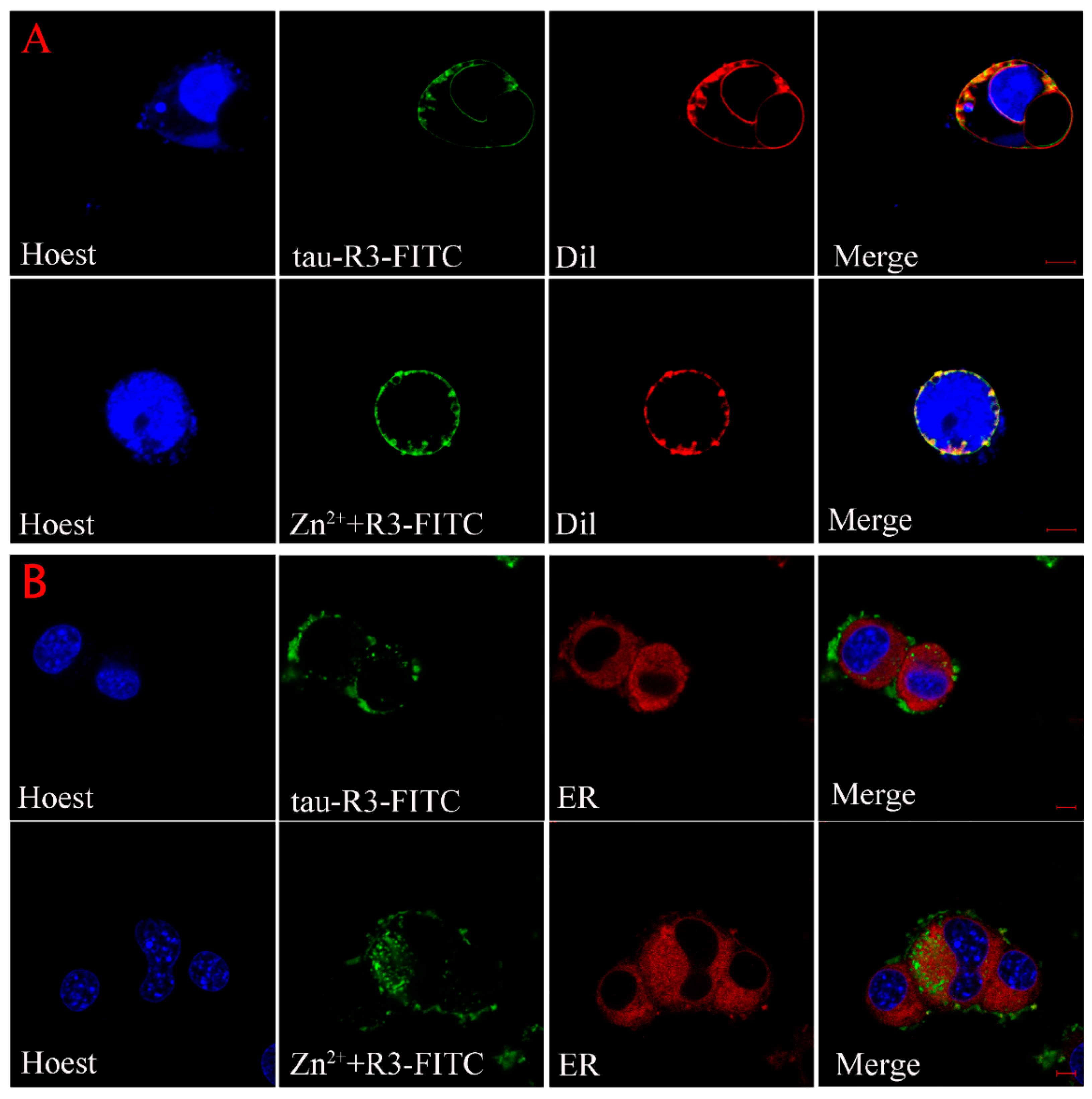
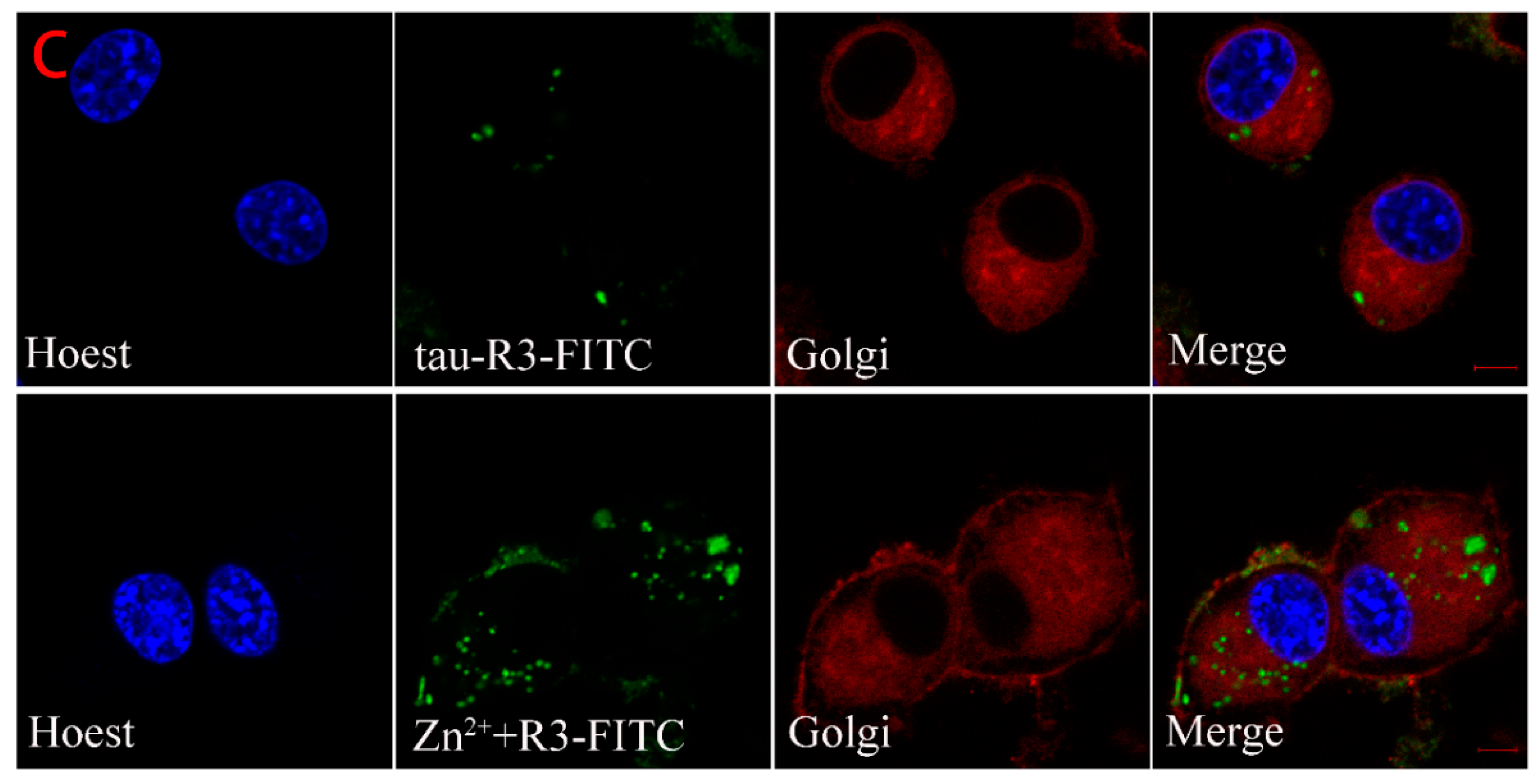
2.6. Subcellular Distribution of Tau-R3
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isothermal Titration Calorimetry (ITC)
4.3. UV-Vis Spectroscopy
4.4. Monitoring Tau-R3 Fibrillization by Thioflavin T Fluorescence
4.5. Circular Dichroism Spectroscopy
4.6. Atomic Force Microscope (AFM)
4.7. Cell Culture
4.8. Cell Viability Measurement
4.9. Measurement of ROS
4.10. Neuronal Culture
4.11. Immunofluorescence Analysis of Map2 Protein
4.12. Cellular Localization of Tau-R3 and Zn2+-Tau-R3 in N2A Cells
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| NFT | neurofibrillary tangles |
| tau-R3 | the third repeat unit of the microtubule-binding domain of tau |
| ROS | reactive oxygen species |
| PP2A | protein phosphatase 2A |
| Aβ | amyloid-β |
| p-tau | hyperphosphorylated tau |
| MAP | microtubule-associated protein |
| PHF | paired helix filaments |
| ITC | isothermal titration calorimetry |
| ThT | thioflavin T |
| CD | circular dichroism spectroscopy |
| FFT | Fourier transform filter |
| DCFH-DA | 2,7-Dichlorodi-hydrofluorescein diacetate |
| PB | Phosphate buffer |
| FITC | fluoresceine isothiocyanate |
| DMEM | dulbecco’s modified eagle medium |
| PBS | phosphate buffer saline |
References
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and his disease: A brief history. Neurol. Sci. 2011, 32, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G. Reprint of “Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein”. Biochem. Biophysic. Res. Commun. 2012, 425, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- World Alzheimer Report 2018. Available online: https://www.sogou.com/link?url=hedJjaC291MORyDunQ5_N0zLsl91bt5WDjCKgfruEPPVigbO6Jra9wOvti-wiKWCreaVHN88tHY (accessed on 21 September 2018).
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. Differential localization of MAP-2 and tau in mammalian neurons in situ. Ann. N. Y. Acad. Sci. 1986, 466, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Wee, M.; Chegini, F.; Power, J.H.T.; Majd, S. Tau Positive Neurons Show Marked Mitochondrial Loss and Nuclear Degradation in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 928–937. [Google Scholar] [CrossRef]
- Liu, F.; Gong, C.X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef]
- Wille, H.; Drewes, G.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J. Cell Biol. 1992, 118, 573–584. [Google Scholar] [CrossRef] [Green Version]
- von Bergen, M.; Barghorn, S.; Li, L.; Marx, A.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biolog. Chem. 2001, 276, 48165–48174. [Google Scholar] [CrossRef]
- Tomoo, K.; Yao, T.M.; Minoura, K.; Hiraoka, S.; Sumida, M.; Taniguchi, T.; Ishida, T. Possible role of each repeat structure of the microtubule-binding domain of the tau protein in in vitro aggregation. J. Biochem. 2005, 138, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. The metal theory of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S277–S281. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. The relevance of metals in the pathophysiology of neurodegeneration, pathological considerations. Int. Rev. Neurobiol. 2013, 110, 1–47. [Google Scholar] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurolog. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, Y.; Shang, Y.; Gu, X.; Zhu, Y.; Zhu, L. NBD-BPEA regulates Zn(2+)- or Cu(2+)-induced Abeta40 aggregation and cytotoxicity. Food Chem. Toxicol. 2018, 119, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Kolandaivel, P. Fe(2+) binding on amyloid beta-peptide promotes aggregation. Proteins 2016, 84, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.F.; Li, Y.M.; Du, J.T.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y.F. Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers 2005, 79, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zheng, Y.; Wang, Z.; Chen, Y.; Zhou, R.; Song, G.; Ni, J.; Liu, Q. Inhibitory act of selenoprotein P on Cu(+)/Cu(2+)-induced tau aggregation and neurotoxicity. Inorg. Chem. 2014, 53, 11221–11230. [Google Scholar] [CrossRef]
- Prasad, A.S. Impact of the discovery of human zinc deficiency on health. J. Am. Coll. Nutr. 2009, 28, 257–265. [Google Scholar] [CrossRef]
- Chasapis, C.T.; Loutsidou, A.C.; Spiliopoulou, C.A.; Stefanidou, M.E. Zinc and human health: An update. Arch. Toxicol. 2012, 86, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Tyszka-Czochara, M.; Grzywacz, A.; Gdula-Argasinska, J.; Librowski, T.; Wilinski, B.; Opoka, W. The role of zinc in the pathogenesis and treatment of central nervous system (CNS) diseases. Implications of zinc homeostasis for proper CNS function. Acta Pol. Pharm. 2014, 71, 369–377. [Google Scholar] [PubMed]
- Beyer, N.; Coulson, D.T.; Heggarty, S.; Ravid, R.; Hellemans, J.; Irvine, G.B.; Johnston, J.A. Zinc transporter mRNA levels in Alzheimer’s disease postmortem brain. J. Alzheimer’s Dis. 2012, 29, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wang, T.; Zheng, W.; Zhao, B.L.; Danscher, G.; Chen, Y.H.; Wang, Z.Y. Zinc overload enhances APP cleavage and Abeta deposition in the Alzheimer mouse brain. PLoS ONE 2010, 5, e15349. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yao, Y.; Lin, J.; Ye, Y.H.; Sun, W.Y.; Tang Dagger, W.X. The solution structure of rat Abeta-(1-28) and its interaction with zinc ion: Insights into the scarcity of amyloid deposition in aged rat brain. J. Biol. Inorg. Chem. 2004, 9, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.Y.; Zhu, Y.Z.; Zhu, H.L.; Fan, J.B.; Chen, J.; Liang, Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 2009, 284, 34648–34657. [Google Scholar] [CrossRef]
- Sun, X.Y.; Wei, Y.P.; Xiong, Y.; Wang, X.C.; Xie, A.J.; Wang, X.L.; Yang, Y.; Wang, Q.; Lu, Y.M.; Liu, R.; et al. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J. Biolog. Chem. 2012, 287, 11174–11182. [Google Scholar] [CrossRef]
- Paglia, G.; Miedico, O.; Cristofano, A.; Vitale, M.; Angiolillo, A.; Chiaravalle, A.E.; Corso, G.; Di Costanzo, A. Distinctive Pattern of Serum Elements During the Progression of Alzheimer’s Disease. Sci. Rep. 2016, 6, 22769. [Google Scholar] [CrossRef] [Green Version]
- Krezel, A.; Maret, W. Dual nanomolar and picomolar Zn(II) binding properties of metallothionein. J. Am. Chem. Soc. 2007, 129, 10911–10921. [Google Scholar] [CrossRef]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Analyt. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- LeVine, H., 3rd. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef]
- Smart, T.G.; Hosie, A.M.; Miller, P.S. Zn2+ ions: Modulators of excitatory and inhibitory synaptic activity. Neuroscientist 2004, 10, 432–442. [Google Scholar] [CrossRef]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef]
- Karakas, E.; Simorowski, N.; Furukawa, H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009, 28, 3910–3920. [Google Scholar] [CrossRef] [Green Version]
- Bitanihirwe, B.K.; Cunningham, M.G. Zinc: The brain’s dark horse. Synapse 2009, 63, 1029–1049. [Google Scholar] [CrossRef]
- Yuan, Y.; Niu, F.; Liu, Y.; Lu, N. Zinc and its effects on oxidative stress in Alzheimer’s disease. Neurol. Sci. 2014, 35, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Jiji, A.C.; Arshad, A.; Dhanya, S.R.; Shabana, P.S.; Mehjubin, C.K.; Vijayan, V. Zn(2+) Interrupts R4-R3 Association Leading to Accelerated Aggregation of Tau Protein. Chemistry 2017, 23, 16976–16979. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, H.; Wang, Z.; Qiu, S.; Liu, Q.; Ni, J. Selenoprotein P and selenoprotein M block Zn2+ -mediated Abeta42 aggregation and toxicity. Metallomics 2013, 5, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.Y.; Zhang, D.L.; Liu, X.L.; Li, X.S.; Cheng, X.Q.; Chen, J.; Du, H.N.; Liang, Y. Pathological concentration of zinc dramatically accelerates abnormal aggregation of full-length human Tau and thereby significantly increases Tau toxicity in neuronal cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.Y.; Devred, F.; Byrne, D.; La Rocca, R.; Ninkina, N.N.; Peyrot, V.; Tsvetkov, P.O. Zinc Induces Temperature-Dependent Reversible Self-Assembly of Tau. J. Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Wang, Z.; Zheng, Y.; Li, H.; Ni, J.; Liu, Q. Inhibitory effect of selenoprotein P on Cu(+)/Cu(2+)-induced Abeta42 aggregation and toxicity. Inorg. Chem. 2014, 53, 1672–1678. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Du, X.; Ni, J. Zn2+ Aggravates Tau Aggregation and Neurotoxicity. Int. J. Mol. Sci. 2019, 20, 487. https://doi.org/10.3390/ijms20030487
Li X, Du X, Ni J. Zn2+ Aggravates Tau Aggregation and Neurotoxicity. International Journal of Molecular Sciences. 2019; 20(3):487. https://doi.org/10.3390/ijms20030487
Chicago/Turabian StyleLi, Xuexia, Xiubo Du, and Jiazuan Ni. 2019. "Zn2+ Aggravates Tau Aggregation and Neurotoxicity" International Journal of Molecular Sciences 20, no. 3: 487. https://doi.org/10.3390/ijms20030487
APA StyleLi, X., Du, X., & Ni, J. (2019). Zn2+ Aggravates Tau Aggregation and Neurotoxicity. International Journal of Molecular Sciences, 20(3), 487. https://doi.org/10.3390/ijms20030487