mTOR Inhibitors in Advanced Biliary Tract Cancers


Abstract
:1. Introduction of Bile Duct Cancers
2. Current Evidences of Systemic Treatment for Advanced Bile Duct Cancers
2.1. In the Era of Chemotherapy
2.2. Development of Targeted Therapy in Advanced BTC
2.3. Immune Checkpoints Inhibitors
3. Molecular Alterations in Cholangiocarcinoma
4. mTOR Pathway in Cancers
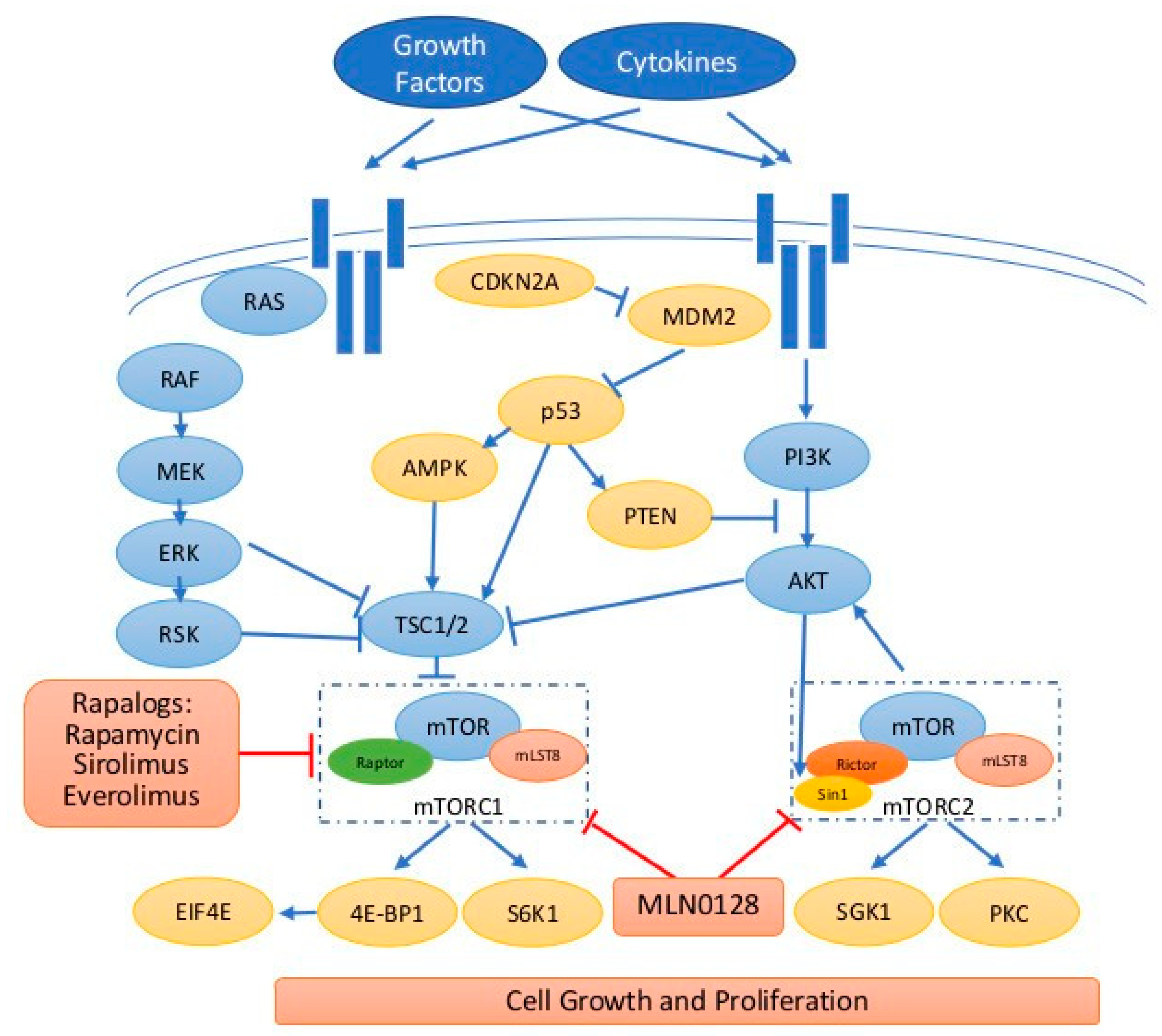
4.1. mTOR, Its Complexes and Downstream Regulations in Cancers
4.2. Upstream Regulation of mTOR in Cancers
4.2.1. The Physiological Regulation of mTOR Pathway
4.2.2. Alterations of mTOR Pathway in Cancers
5. mTOR Inhibitors
5.1. Rapalogs, First-Generation of mTOR Inhibitors
5.2. Second-Generation mTOR Inhibitors
6. Sustained mTORC1/2 Signaling Activation as a Driver of Resistance to Anti-Cancer Treatment
7. Preclinical Studies of mTOR Inhibitors in BTC
7.1. The Rationale of mTOR Inhibitors Alone or in Combination with Chemotherapeutic Agents in Cholangiocarcinoma
7.2. Preclinical Studies of Rapalogs in BTC
7.3. New Generation mTOR Inhibitors in BTC
7.4. Dual PI3K/mTOR Inhibitors in BTC
7.5. Other Indirect Inhibition of mTOR Pathway
8. mTOR Inhibitors in Clinical Setting
8.1. Clinical Studies of Everolimus in Advanced BTC
8.2. Clinical Studies of Sirolimus in Advanced BTC
8.3. Clinical Studies of New Generation mTOR Inhibitors in Advanced BTC
9. Summary of mTOR Inhibitors in BTC
Funding
Conflicts of Interest
References
- Patel, T. Cholangiocarcinoma. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 33–42. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Patel, T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001, 33, 1353–1357. [Google Scholar] [CrossRef] [Green Version]
- Shaib, Y.H.; Davila, J.A.; McGlynn, K.; El-Serag, H.B. Rising incidence of intrahepatic cholangiocarcinoma in the United States: A true increase? J. Hepatol. 2004, 40, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Jarnagin, W.R.; Fong, Y.; DeMatteo, R.P.; Gonen, M.; Burke, E.C.; Bodniewicz, B.J.; Youssef, B.M.; Klimstra, D.; Blumgart, L.H. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann. Surg. 2001, 234, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Wasan, H.; Johnson, P.; Jones, E.; Dixon, L.; Swindell, R.; Baka, S.; Maraveyas, A.; Corrie, P.; Falk, S.; et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: A multicentre randomised phase II study—The UK ABC-01 Study. Br. J. Cancer 2009, 101, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Glimelius, B.; Hoffman, K.; Sjoden, P.O.; Jacobsson, G.; Sellstrom, H.; Enander, L.K.; Linne, T.; Svensson, C. Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann. Oncol. 1996, 7, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Eckel, F.; Schmid, R.M. Chemotherapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Br. J. Cancer 2007, 96, 896–902. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Wu, C.E.; Hsu, H.C.; Shen, W.C.; Lin, Y.C.; Wang, H.M.; Chang, J.W.; Chen, J.S. Chemotherapy with gemcitabine plus cisplatin in patients with advanced biliary tract carcinoma at Chang Gung Memorial Hospital: A retrospective analysis. Chang Gung Med. J. 2012, 35, 420–427. [Google Scholar] [PubMed]
- Ueno, M.; Morizane, C.; Okusaka, T.; Mizusawa, J.; Katayama, H.; Ikeda, M. Randomized phase III study of gemcitabine plus S-1 combination therapy versus gemcitabine plus cisplatin combination therapy in advanced biliary tract cancer: A Japan Clinical Oncology Group study (JCOG1113, FUGA-BT). J. Clin. Oncol. 2018, 36, 205. [Google Scholar] [CrossRef]
- Zhu, A.X.; Meyerhardt, J.A.; Blaszkowsky, L.S.; Kambadakone, A.; Muzikansky, A.; Zheng, H.; Clark, J.W.; Abrams, T.A.; Chan, J.A.; Enzinger, P.C.; et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: A phase 2 study. Lancet Oncol. 2010, 11, 48–54. [Google Scholar] [CrossRef]
- Rubovszky, G.; Lang, I.; Ganofszky, E.; Horvath, Z.; Juhos, E.; Nagy, T.; Szabo, E.; Szentirmay, Z.; Budai, B.; Hitre, E. Cetuximab, gemcitabine and capecitabine in patients with inoperable biliary tract cancer: A phase 2 study. Eur. J. Cancer 2013, 49, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; de la Fouchardiere, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.F.; Viret, F.; et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, C.; Chiang, N.J.; Tsai, C.S.; Tsou, H.H.; Huang, S.F.; Bai, L.Y.; Chang, I.C.; Shiah, H.S.; Ho, C.L.; et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann. Oncol. 2015, 26, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Eckel, F.; Schmid, R.M. Chemotherapy and targeted therapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Chemotherapy 2014, 60, 13–23. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Bang, Y.J.; Doi, T.; De Braud, F.; Piha-Paul, S.; Hollebecque, A.; Razak, A.R.A.; Lin, C.C.; Ott, P.A.; He, A.R.; Yuan, S.S.; et al. Safety and efficacy of pembrolizumab (MK-3475) in patients (pts) with advanced biliary tract cancer: Interim results of KEYNOTE-028. Eur. J. Cancer 2015, 51, S112. [Google Scholar] [CrossRef]
- Goldstein, D.; Lemech, C.; Valle, J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017, 2, e000152. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Tsuchida, A.; Iwao, T.; Eguchi, N.; Sasaki, T.; Morinaka, K.; Matsubara, K.; Kawasaki, Y.; Yamamoto, S.; Kajiyama, G. Gene mutations of K-ras in gallbladder mucosae and gallbladder carcinoma with an anomalous junction of the pancreaticobiliary duct. Am. J. Gastroenterol. 1999, 94, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.T.; Kim, J.; Jang, Y.H.; Lee, W.J.; Ryu, J.K.; Park, Y.K.; Kim, S.W.; Kim, W.H.; Yoon, Y.B.; Kim, C.Y. Genetic alterations in gallbladder adenoma, dysplasia and carcinoma. Cancer Lett. 2001, 169, 59–68. [Google Scholar] [CrossRef]
- Tannapfel, A.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Kockerling, F.; Hauss, J.; Wittekind, C. Frequency of p16(INK4A) alterations and k-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut 2000, 47, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Witzigmann, H.; Hauss, J.; Wittekind, C. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 2003, 52, 706–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin. Cancer Res. 2006, 12, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Riener, M.O.; Bawohl, M.; Clavien, P.A.; Jochum, W. Rare PIK3CA hotspot mutations in carcinomas of the biliary tract. Genes Chromosomes Cancer 2008, 47, 363–367. [Google Scholar] [CrossRef]
- Argani, P.; Shaukat, A.; Kaushal, M.; Wilentz, R.E.; Su, G.H.; Sohn, T.A.; Yeo, G.J.; Cameron, J.L.; Kern, S.E.; Hruban, R.H. Differing rates of loss of Dpc4 expression and of p53 overexpression among carcinomas of the proximal and distal bile ducts—Evidence for biologic distinction. Cancer 2001, 91, 1332–1341. [Google Scholar] [CrossRef]
- Ueki, T.; Hsing, A.W.; Gao, Y.T.; Wang, B.S.; Shen, M.C.; Cheng, J.; Deng, J.; Fraumeni, J.F., Jr.; Rashid, A. Alterations of p16 and prognosis in biliary tract cancers from a population-based study in China. Clin. Cancer Res. 2004, 10, 1717–1725. [Google Scholar] [CrossRef]
- Hezel, A.F.; Deshpande, V.; Zhu, A.X. Genetics of biliary tract cancers and emerging targeted therapies. J. Clin. Oncol. 2010, 28, 3531–3540. [Google Scholar] [CrossRef]
- Ekshyyan, O.; Anandharaj, A.; Nathan, C.A.O. Dual PI3K/mTOR Inhibitors: Does p53 Modulate Response? Clin. Cancer Res. 2013, 19, 3719–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, D.E.; Rahman, A.; Hidalgo, M.; Thuluvath, P.J.; Lillemoe, K.D.; Schulick, R.; Ku, J.L.; Park, J.G.; Miyazaki, K.; Ashfaq, R.; et al. Identification of novel cellular targets in biliary tract cancers using global gene expression technology. Am. J. Pathol. 2003, 163, 217–229. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B. The mTOR signalling cascade: Paving new roads to cure neurological disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, W.; Hu, X.; Dorrance, A.; Garzon, R.; Houghton, P.J.; Shen, C. Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Sci. Rep. 2017, 7, 1535. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Hong, S.M.; Choi, B.Y.; Cho, H.; Yu, E.; Hewitt, S.M. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin. Cancer Res. 2009, 15, 660–667. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Huang, S.L. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin. J. Cancer 2012, 31, 8–18. [Google Scholar] [CrossRef]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Gene Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.X.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. T 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Schulick, R. Identification of Novel Cellular Targets in Biliary Tract Cancers Using Global Gene Expression Technology (vol 163, pg 217, 2003). Am. J. Pathol. 2017, 187, 936. [Google Scholar]
- Markman, B.; Dienstmann, R.; Tabernero, J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget 2010, 1, 530–543. [Google Scholar]
- Wang, X.M.; Proud, C.G. mTORC2 is a tyrosine kinase. Cell Res. 2016, 26, 1–2. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR Complex 2 Is Required for the Development of Prostate Cancer Induced by Pten Loss in Mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guri, Y.; Hall, M.N. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer 2016, 2, 688–697. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Khuri, F.R. Targeting the PI3K/AKT/mTOR pathway: Biomarkers of success and tribulation. Am. Soc. Clin. Oncol. Educ. Book 2013. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Waldman, T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 2010, 347, 21–41. [Google Scholar] [PubMed]
- Whale, A.D.; Colman, L.; Lensun, L.; Rogers, H.L.; Shuttleworth, S.J. Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Signal Transduct. Target. Ther. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, M.P. PIP2 and PIP3: Complex roles at the cell surface. Cell 2000, 100, 603–606. [Google Scholar] [CrossRef]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Roa, I.; Garcia, H.; Game, A.; de Toro, G.; de Aretxabala, X.; Javle, M. Somatic Mutations of PI3K in Early and Advanced Gallbladder Cancer Additional Options for an Orphan Cancer. J. Mol. Diagn. 2016, 18, 388–394. [Google Scholar] [CrossRef]
- Petzold, J.; Lederer, E.; Reihs, R.; Ernst, C.; Bettermann, K.; Halbwedl, I.; Lax, S.; Park, Y.N.; Kim, K.S.; Kiesslich, T.; et al. Pten inactivation and alteration of the pi3k/AKT/MTOR pathway in biliary tract cancer. Anticancer Res. 2014, 34, 5942–5943. [Google Scholar]
- Kornprat, P.; Rehak, P.; Ruschoff, J.; Langner, C. Expression of IGF-I, IGF-II, and IGF-IR in gallbladder carcinoma. A systematic analysis including primary and corresponding metastatic tumours. J. Clin. Pathol. 2006, 59, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.; Lorenz, J.; Mossner, J.; Wiedmann, M. Treatment of biliary tract cancer with NVP-AEW541: Mechanisms of action and resistance. World J. Gastroenterol. 2010, 16, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. The Pharmacology of mTOR Inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilella-Bach, M.; Nuzzi, P.; Fang, Y.; Chen, J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. J. Biol. Chem. 1999, 274, 4266–4272. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar] [CrossRef]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Mao, J.H.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D.; Yi, F.; Ji, D.M. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef]
- Liu, Q.S.; Kirubakaran, S.; Hur, W.; Niepel, M.; Westover, K.; Thoreen, C.C.; Wang, J.H.; Ni, J.; Patricelli, M.P.; Vogel, K.; et al. Kinome-wide Selectivity Profiling of ATP-competitive Mammalian Target of Rapamycin (mTOR) Inhibitors and Characterization of Their Binding Kinetics. J. Biol. Chem. 2012, 287, 9742–9752. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Wheater, M.; Din, O.; Geldart, T.; Boleti, E.; Stockdale, A.; Sundar, S.; Robinson, A.; Ahmed, I.; Wimalasingham, A.; et al. A Randomised Phase 2 Study of AZD2014 Versus Everolimus in Patients with VEGF-Refractory Metastatic Clear Cell Renal Cancer. Eur. Urol. 2016, 69, 450–456. [Google Scholar] [CrossRef]
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.S.; Chen, S.A. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, T.; Beyens, M.; de Beeck, K.O.; Dogan, F.; van Koetsveld, P.M.; Pauwels, P.; Mortier, G.; Vangestel, C.; de Herder, W.; Van Camp, G.; et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer 2016, 114, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Ferreira, M.; Dubey, S.; Zaiss, M.; Harper-Wynne, C.; Makris, A.; Brown, V.; Kristeleit, H.; Patel, G.; Perello, A.; et al. MANTA: A randomized phase II study of fulvestrant in combination with the dual mTOR inhibitor AZD2014 or everolimus or fulvestrant alone in estrogen receptor-positive advanced or metastatic breast cancer. Cancer Res. 2018, 78. Abstract GS2-07. [Google Scholar] [CrossRef]
- Kelsey, I.; Manning, B.D. mTORC1 status dictates tumor response to targeted therapeutics. Sci. Signal. 2013, 6, pe31. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, E.; Manning, B.D. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. Role of mTOR in anticancer drug resistance: Perspectives for improved drug treatment. Drug Resist. Updates 2008, 11, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, K.; Kobayashi, A.; Motoyama, H.; Kitazawa, M.; Shimizu, A.; Notake, T.; Yokoyama, T.; Matsumura, T.; Takeoka, M.; Miyagawa, S.I. Survival pathway of cholangiocarcinoma via AKT/mTOR signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib. Oncol. Rep. 2018, 39, 843–850. [Google Scholar] [CrossRef]
- Li, Q.; Xia, X.; Ji, J.; Ma, J.; Tao, L.; Mo, L.; Chen, W. MiR-199a-3p enhances cisplatin sensitivity of cholangiocarcinoma cells by inhibiting mTOR signaling pathway and expression of MDR1. Oncotarget 2017, 8, 33621–33630. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Feng, T.; Ke, Q.; Fan, N.; Li, L.; Li, Z.; Dong, C.; Wang, C.; Xu, F.; Li, Y.; et al. Metformin inhibits proliferation and enhances chemosensitivity of intrahepatic cholangiocarcinoma cell lines. Oncol. Rep. 2014, 31, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Wandee, J.; Prawan, A.; Senggunprai, L.; Kongpetch, S.; Tusskorn, O.; Kukongviriyapan, V. Metformin enhances cisplatin induced inhibition of cholangiocarcinoma cells via AMPK-mTOR pathway. Life Sci. 2018, 207, 172–183. [Google Scholar] [CrossRef]
- Ahn, C.S.; Han, J.A.; Lee, H.S.; Lee, S.; Pai, H.S. The PP2A regulatory subunit Tap46, a component of the TOR signaling pathway, modulates growth and metabolism in plants. Plant Cell 2011, 23, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.C.; Han, D.D.; Li, X.L.; Ma, J.; Wu, Q.; Dong, H.M.; Bai, C.; He, Q. Fyn knockdown inhibits migration and invasion in cholangiocarcinoma through the activated AMPK/mTOR signaling pathway. Oncol. Lett. 2018, 15, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Moolthiya, P.; Tohtong, R.; Keeratichamroen, S.; Leelawat, K. Role of mTOR inhibitor in cholangiocarcinoma cell progression. Oncol. Lett. 2014, 7, 854–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heits, N.; Heinze, T.; Bernsmeier, A.; Kerber, J.; Hauser, C.; Becker, T.; Kalthoff, H.; Egberts, J.H.; Braun, F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer 2016, 16, 322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.F.; Zhang, C.Y.; Zhou, H.; Xiao, B.; Cheng, Y.; Wang, J.J.; Yao, F.L.; Duan, C.Y.; Chen, R.; Liu, Y.P.; et al. Synergistic antitumor activity of the combination of salubrinal and rapamycin against human cholangiocarcinoma cells. Oncotarget 2016, 7, 85492–85501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.G.; Lin, K.J.; Wang, F.; Chen, T.C.; Yen, T.C.; Yeh, T.S. Synergistic antiproliferative effects of an mTOR inhibitor (rad001) plus gemcitabine on cholangiocarcinoma by decreasing choline kinase activity. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Song, X.H.; Cao, D.; Xu, Z.; Fan, B.A.; Che, L.; Hu, J.J.; Chen, B.; Dong, M.J.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef]
- Song, X.; Liu, X.; Wang, H.; Wang, J.; Qiao, Y.; Cigliano, A.; Utpatel, K.; Ribback, S.; Pilo, M.G.; Serra, M.; et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Fraveto, A.; Cardinale, V.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Grazi, G.L.; Napoletano, C.; Semeraro, R.; Lustri, A.M.; Costantini, D.; et al. Sensitivity of Human Intrahepatic Cholangiocarcinoma Subtypes to Chemotherapeutics and Molecular Targeted Agents: A Study on Primary Cell Cultures. PLoS ONE 2015, 10, e0142124. [Google Scholar] [CrossRef]
- Chen, M.H.; Chiang, K.C.; Cheng, C.T.; Huang, S.C.; Chen, Y.Y.; Chen, T.W.; Yeh, T.S.; Jan, Y.Y.; Wang, H.M.; Weng, J.J.; et al. Antitumor activity of the combination of an HSP90 inhibitor and a PI3K/mTOR dual inhibitor against cholangiocarcinoma. Oncotarget 2014, 5, 2372–2389. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhang, Q.Y.; Li, J.L.; Zhang, N.; Hua, Y.P.; Xu, L.X.; Deng, Y.B.; Lai, J.M.; Peng, Z.W.; Peng, B.G.; et al. Apatinib inhibits VEGF signaling and promotes apoptosis in intrahepatic cholangiocarcinoma. Oncotarget 2016, 7, 17220–17229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.E.; Koay, T.S.; Esfandiari, A.; Ho, Y.H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, 3. [Google Scholar] [CrossRef]
- Andersen, N.J.; Boguslawski, E.B.; Kuk, C.Y.; Chambers, C.M.; Duesbery, N.S. Combined inhibition of MEK and mTOR has a synergic effect on angiosarcoma tumorgrafts. Int. J. Oncol. 2015, 47, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.E.; Esfandiari, A.; Ho, Y.H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Laroche, A.; Chaire, V.; Algeo, M.P.; Karanian, M.; Fourneaux, B.; Italiano, A. MDM2 antagonists synergize with PI3K/mTOR inhibition in well-differentiated/dedifferentiated liposarcomas. Oncotarget 2017, 8, 53968–53977. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.L.; Wang, M.M.; Tong, E.J.; Sun, J.; Li, M.; Miao, Z.B.; Li, Y.L.; Zhu, B.H.; Xu, J.J. Benefit of everolimus in treatment of an intrahepatic cholangiocarcinoma patient with a PIK3CA mutation. World J. Gastroenterol. 2017, 23, 4311–4316. [Google Scholar] [CrossRef] [PubMed]
- Verzoni, E.; Pusceddu, S.; Buzzoni, R.; Garanzini, E.; Damato, A.; Biondani, P.; Testa, I.; Grassi, P.; Bajetta, E.; DeBraud, F.; et al. Safety profile and treatment response of everolimus in different solid tumors: An observational study. Future Oncol. 2014, 10, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Buzzoni, R.; Pusceddu, S.; Bajetta, E.; De Braud, F.; Platania, M.; Iannacone, C.; Cantore, M.; Mambrini, A.; Bertolini, A.; Alabiso, O.; et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: A phase II ITMO study. Ann. Oncol. 2014, 25, 1597–1603. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Kang, W.K.; Lim, H.Y. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer 2017, 17, 211. [Google Scholar] [CrossRef]
- Lau, D.K.; Tay, R.Y.; Yeung, Y.H.; Chionh, F.; Mooi, J.; Murone, C.; Skrinos, E.; Price, T.J.; Mariadason, J.M.; Tebbutt, N.C. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: The RADiChol Study. Br. J. Cancer 2018, 118, 966–971. [Google Scholar] [CrossRef]
- Rizell, M.; Andersson, M.; Cahlin, C.; Hafstrom, L.; Olausson, M.; Lindner, P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int. J. Clin. Oncol. 2008, 13, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.S.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, S.T. Pilot study of sirolimus in patients with PIK3CA mutant/amplified refractory solid cancer. Mol. Clin. Oncol. 2017, 7, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, B.A.; Borad, M.J.; Qi, Y.; Kim, G.P.; Northfelt, D.W.; Erlichman, C.; Alberts, S.R. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Investig. New Drugs 2014, 32, 710–716. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Compound(s) | Phase | Patients | Response | Survival |
|---|---|---|---|---|
| Everolimus, 1 L [106] | Case report | iCCA (n = 1) with PIK3CA mutation | PR | PFS > 6 m |
| Everolimus [107] | Phase I | Advanced BTC (n = 22) | DCR: 50% (11/22) | NA |
| Everolimus (>2 L) [108] | Phase II | Advanced BTC (n = 39) | DCR: 44.7% RR: 5.1% (including 1 CR) | mPFS: 3.2 m (1.8–4.0) mOS: 7.7 m (5.5–13.2) |
| Everolimus [109] | Phase II | CCA (n = 1), PTEN loss | SD | NA |
| Everolimus (1 L) [110] | Phase II | Advanced BTC (n = 27) | DCR at 12 weeks: 48% PR: 12% (3/25) SD: 60% (15/25) | mPFS: 5.5 m (2.2–10.0) mOS: 9.5 m (5.5–16.6) |
| Sirolimus [111] | Phase II | iCCA (n = 9) | SD: 33% (3/9) PD: 67% (6/9) | mOS:7 (2.6–35) |
| Sirolimus [112] | Phase II | hilar CCA (n = 1) with PIK3CA mutation | PD | PFS: 0.9 m |
| Everolimus, gemcitabine, cisplatin (1 L) [113] | Phase I | Cohort III, CCA and GBC (n = 10) | SD: 60% (6/10) PD: 40% (4/10) | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-E.; Chen, M.-H.; Yeh, C.-N. mTOR Inhibitors in Advanced Biliary Tract Cancers. Int. J. Mol. Sci. 2019, 20, 500. https://doi.org/10.3390/ijms20030500
Wu C-E, Chen M-H, Yeh C-N. mTOR Inhibitors in Advanced Biliary Tract Cancers. International Journal of Molecular Sciences. 2019; 20(3):500. https://doi.org/10.3390/ijms20030500
Chicago/Turabian StyleWu, Chao-En, Ming-Huang Chen, and Chun-Nan Yeh. 2019. "mTOR Inhibitors in Advanced Biliary Tract Cancers" International Journal of Molecular Sciences 20, no. 3: 500. https://doi.org/10.3390/ijms20030500
APA StyleWu, C. -E., Chen, M. -H., & Yeh, C. -N. (2019). mTOR Inhibitors in Advanced Biliary Tract Cancers. International Journal of Molecular Sciences, 20(3), 500. https://doi.org/10.3390/ijms20030500