Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases




Abstract
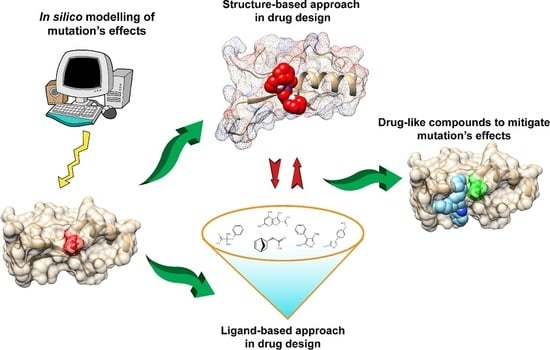
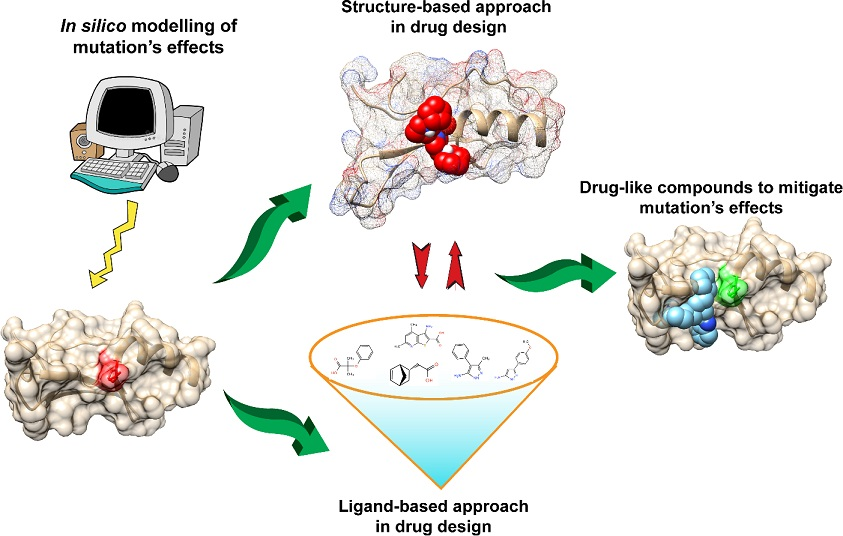
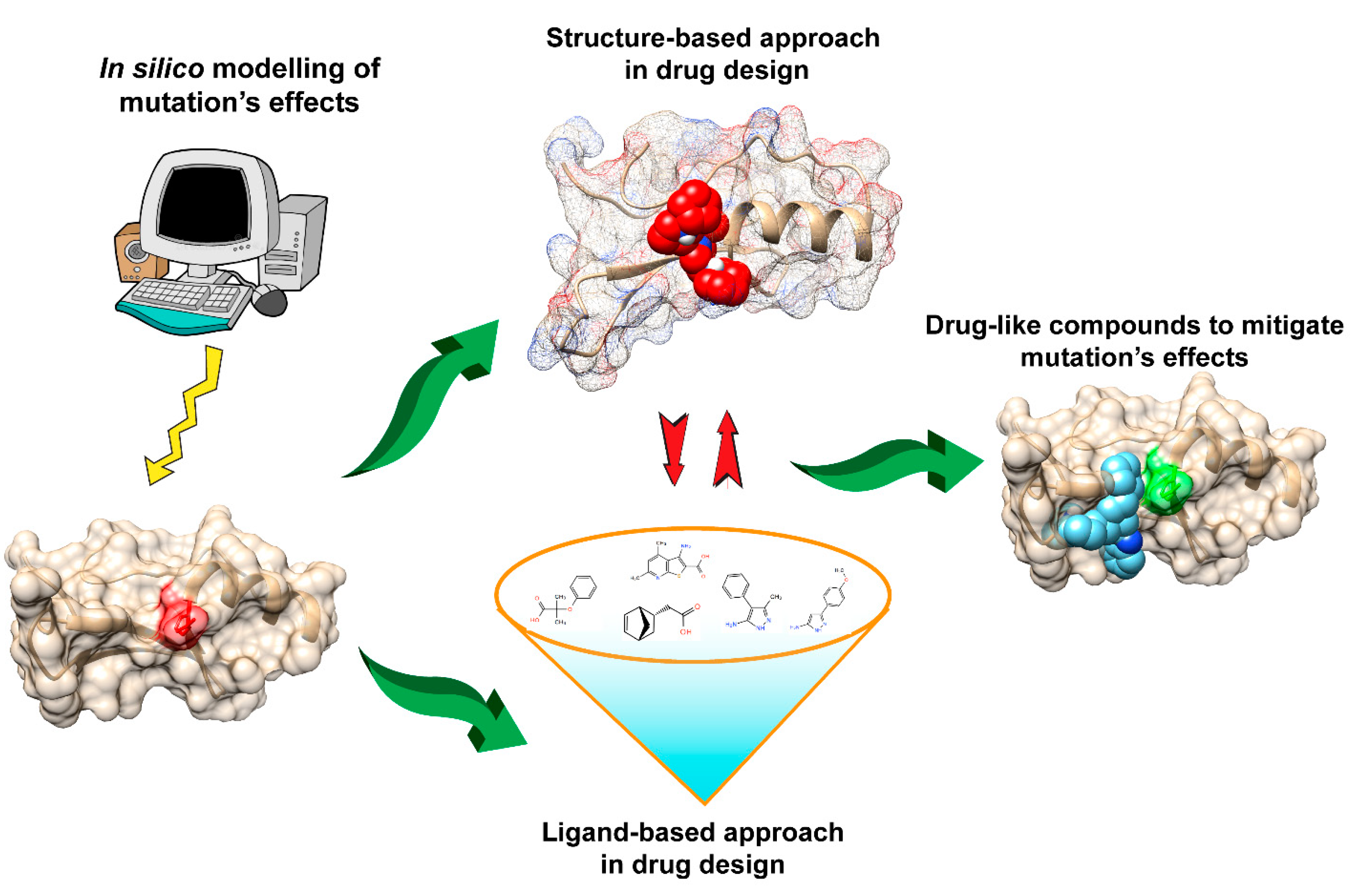
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Effect of Mutations on Stability and Binding
2. Mutation and its Compensation: Structural Plasticity and Conformational Relaxation
3. Mutations in IDPs as Compared to Globular and Membrane Proteins
4. Probing the Role of Mutations in Diseases: Tracking Changes in Thermodynamic Parameters
5. Statistical Classification of Mutations Based on Their Degree of Harmfulness
6. Mitigating and Clustering the Effects of Disease-Causing Genetic Variants in Relation to Drug Design
7. Structure-Based Approach in Drug Design
7.1. Docking
7.2. Structure-Based Pharmacophore Design
8. Ligand-Based Approaches in Drug Design
9. Aiding Drug Design by the Knowledge of Mutations on Globular, Membrane and Disordered Proteins
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shoichet, B.K.; Baase, W.A.; Kuroki, R.; Matthews, B.W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA 1995, 92, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hawkins, P.N.; Booth, D.R.; Vigushin, D.M.; Tennent, G.A.; Soutar, A.K.; Totty, N.; Nguyen, O.; Blake, C.C.F.; Terry, C.J.; et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993, 362, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Hartley, R.W. Directed mutagenesis and barnase-barstar recognition. Biochemistry 1993, 32, 5978–5984. [Google Scholar] [CrossRef] [PubMed]
- Buckle, A.M.; Schreiber, G.; Fersht, A.R. Protein-protein recognition: Crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry 1994, 33, 8878–8889. [Google Scholar] [CrossRef]
- Schreiber, G.; Fersht, A.R. Energetics of protein-protein interactions: Analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995, 248, 478–486. [Google Scholar] [CrossRef]
- Wang, T.; Tomic, S.; Gabdoulline, R.R.; Wade, R.C. How optimal are the binding energetics of barnase and barstar? Biophys. J. 2004, 87, 1618–1630. [Google Scholar] [CrossRef] [PubMed]
- Spång, H.C.L.; Braathen, R.; Bogen, B. Heterodimeric Barnase-Barstar Vaccine Molecules: Influence of One versus Two Targeting Units Specific for Antigen Presenting Cells. PLoS ONE 2012, 7, e45393. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M. The interpretation of protein structures: Total volume, group volume distributions and packing density. J. Mol. Biol. 1974, 82, 1–14. [Google Scholar] [CrossRef]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Mapping the distribution of packing topologies within protein interiors shows predominant preference for specific packing motifs. BMC Bioinf. 2011, 12, 195. [Google Scholar] [CrossRef]
- Javadpour, M.M.; Eilers, M.; Groesbeek, M.; Smith, S.O. Helix packing in polytopic membrane proteins: Role of glycine in transmembrane helix association. Biophys. J. 1999, 77, 1609–1618. [Google Scholar] [CrossRef]
- Eilers, M.; Shekar, S.C.; Shieh, T.; Smith, S.O.; Fleming, P.J. Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 5796–5801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Sen, M.; Bhattacharya, D.; Saha, P. The jigsaw puzzle model: Search for conformational specificity in protein interiors. J. Mol. Biol. 2003, 333, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Charneski, C.A.; Hurst, L.D. Positive charge loading at protein termini is due to membrane protein topology, not a translational ramp. Mol. Biol. Evol. 2014, 31, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.A.; Tipper, D.J. The Role of Charged Residues in Determining Transmembrane Protein Insertion Orientation in Yeast. J. Biol. Chem. 1996, 271, 24625–24633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Skach, W.R. Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 2009, 16, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H. Roles of electrostatic interaction in proteins. Q. Rev. Biophys. 1996, 29, 1–90. [Google Scholar] [CrossRef]
- Basu, S.; Biswas, P. Salt-bridge dynamics in intrinsically disordered proteins: A trade-off between electrostatic interactions and structural flexibility. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2018, 1866, 624–641. [Google Scholar] [CrossRef]
- Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Gassner, N.C.; Baase, W.A.; Matthews, B.W. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc. Natl. Acad. Sci. USA 1996, 93, 12155–12158. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Applications of complementarity plot in error detection and structure validation of proteins. Indian J. Biochem. Biophys. 2014, 51, 188–200. [Google Scholar] [PubMed]
- Liang, J.; Naveed, H.; Jimenez-Morales, D.; Adamian, L.; Lin, M. Computational studies of membrane proteins: Models and predictions for biological understanding. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 927–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.S.; Fung, H.K.; Rajgaria, R.; Filizola, M.; Weinstein, H.; Floudas, C.A. Mutations Affecting the Oligomerization Interface of G-Protein-Coupled Receptors Revealed by a Novel De Novo Protein Design Framework. Biophys. J. 2008, 94, 2470–2481. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bowie, J.U. Building a Thermostable Membrane Protein. J. Biol. Chem. 2000, 275, 6975–6979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Situ, A.J.; Ulmer, T.S. Structural and thermodynamic basis of proline-induced transmembrane complex stabilization. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Sepulveda, E.; Hartmann, M.D.; Kogenaru, M.; Ursinus, A.; Sulz, E.; Albrecht, R.; Coles, M.; Martin, J.; Lupas, A.N. Origin of a folded repeat protein from an intrinsically disordered ancestor. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Baruah, A.; Biswas, P. Globular–disorder transition in proteins: A compromise between hydrophobic and electrostatic interactions? Phys. Chem. Chem. Phys. 2016, 18, 23207–23214. [Google Scholar] [CrossRef]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Mas, G.; Hiller, S. Conformational plasticity of molecular chaperones involved in periplasmic and outer membrane protein folding. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef]
- Ikura, M.; Ames, J.B. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: Two ways to promote multifunctionality. Proc. Natl. Acad. Sci. USA 2006, 103, 1159–1164. [Google Scholar] [CrossRef] [Green Version]
- Bastolla, U.; Porto, M.; Roman, H.E. The emerging dynamic view of proteins: Protein plasticity in allostery, evolution and self-assembly. Biochim. Biophys. Acta 2013, 1834, 817–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckle, A.M.; Cramer, P.; Fersht, A.R. Structural and energetic responses to cavity-creating mutations in hydrophobic cores: Observation of a buried water molecule and the hydrophilic nature of such hydrophobic cavities. Biochemistry 1996, 35, 4298–4305. [Google Scholar] [CrossRef]
- Eriksson, A.E.; Baase, W.A.; Zhang, X.J.; Heinz, D.W.; Blaber, M.; Baldwin, E.P.; Matthews, B.W. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 1992, 255, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Axe, D.D.; Foster, N.W.; Fersht, A.R. Active barnase variants with completely random hydrophobic cores. Proc. Natl. Acad. Sci. USA 1996, 93, 5590–5594. [Google Scholar] [CrossRef] [PubMed]
- Dahiyat, B.I.; Sarisky, C.A.; Mayo, S.L. De novo protein design: Towards fully automated sequence selection. J. Mol. Biol. 1997, 273, 789–796. [Google Scholar] [CrossRef]
- Goraj, K.; Renard, A.; Martial, J.A. Synthesis, purification and initial structural characterization of octarellin, a de novo polypeptide modelled on the alpha/beta-barrel proteins. Protein Eng. 1990, 3, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Offredi, F.; Dubail, F.; Kischel, P.; Sarinski, K.; Stern, A.S.; Van de Weerdt, C.; Hoch, J.C.; Prosperi, C.; François, J.M.; Mayo, S.L.; et al. De novo backbone and sequence design of an idealized alpha/beta-barrel protein: Evidence of stable tertiary structure. J. Mol. Biol. 2003, 325, 163–174. [Google Scholar] [CrossRef]
- Teng, S.; Madej, T.; Panchenko, A.; Alexov, E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys. J. 2009, 96, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Kalmar, L.; Tompa, P.; Han, K.-H.; Selenko, P.; Dunker, A.K.; Daughdrill, G.W.; Uversky, V.N. The alphabet of intrinsic disorder. Intrinsically Disord. Proteins 2013, 1, e24360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Söderquist, F.; Wallner, B. Proteus: A random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J. Comput. Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Globular and disordered—The non-identical twins in protein-protein interactions. Front. Mol. Biosci. 2015, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Schymkowitz, J.; Rousseau, F.; Diella, F.; Serrano, L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004, 342, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, J.S.; Miles, A.J.; Araujo, A.P.U.; Wallace, B.A. Differential dehydration effects on globular proteins and intrinsically disordered proteins during film formation. Protein Sci. 2017, 26, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.A.; Singh, V.K.; Jia, Z.; Forman-Kay, J.D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 2006, 15, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, T.R.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Mysterious oligomerization of the amyloidogenic proteins. FEBS J. 2010, 277, 2940–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacic, V.; Iakoucheva, L.M. Disease mutations in disordered regions–exception to the rule? Mol. Biosyst. 2012, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Mechanic, L.E.; Marrogi, A.J.; Welsh, J.A.; Bowman, E.D.; Khan, M.A.; Enewold, L.; Zheng, Y.-L.; Chanock, S.; Shields, P.G.; Harris, C.C. Polymorphisms in XPD and TP53 and mutation in human lung cancer. Carcinogenesis 2005, 26, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 2007, 97, 1–23. [Google Scholar] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [Green Version]
- Feyfant, E.; Sali, A.; Fiser, A. Modeling mutations in protein structures. Protein Sci. 2007, 16, 2030–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, R.A.; Dessailly, B.H.; Orengo, C.A. Residue mutations and their impact on protein structure and function: Detecting beneficial and pathogenic changes. Biochem. J. 2013, 449, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Daudé, D.; Topham, C.M.; Remaud-Siméon, M.; André, I. Probing impact of active site residue mutations on stability and activity of Neisseria polysaccharea amylosucrase. Protein Sci. 2013, 22, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Gerton, J.L.; Ohgi, S.; Olsen, M.; DeRisi, J.; Brown, P.O. Effects of Mutations in Residues near the Active Site of Human Immunodeficiency Virus Type 1 Integrase on Specific Enzyme-Substrate Interactions. J. Virol. 1998, 72, 5046–5055. [Google Scholar]
- Woods, K.N.; Pfeffer, J.; Dutta, A.; Klein-Seetharaman, J. Vibrational resonance, allostery, and activation in rhodopsin-like G protein-coupled receptors. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Luk, L.Y.P.; Javier Ruiz-Pernía, J.; Dawson, W.M.; Roca, M.; Loveridge, E.J.; Glowacki, D.R.; Harvey, J.N.; Mulholland, A.J.; Tuñón, I.; Moliner, V.; et al. Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. USA 2013, 110, 16344–16349. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.; Yi, L.; Gowthaman, R.; Torkamani, A.; Schork, N.J.; Verkhivker, G.M. Sequence and Structure Signatures of Cancer Mutation Hotspots in Protein Kinases. PLoS ONE 2009, 4, e7485. [Google Scholar] [CrossRef]
- Tyukhtenko, S.; Rajarshi, G.; Karageorgos, I.; Zvonok, N.; Gallagher, E.S.; Huang, H.; Vemuri, K.; Hudgens, J.W.; Ma, X.; Nasr, M.L.; et al. Effects of Distal Mutations on the Structure, Dynamics and Catalysis of Human Monoacylglycerol Lipase. Sci. Rep. 2018, 8, 1719. [Google Scholar] [CrossRef] [Green Version]
- Klvaňa, M.; Murphy, D.L.; Jeřábek, P.; Goodman, M.F.; Warshel, A.; Sweasy, J.B.; Florián, J. Catalytic Effects of Mutations of Distant Protein Residues in Human DNA Polymerase β: Theory and Experiment. Biochemistry 2012, 51, 8829–8843. [Google Scholar] [CrossRef] [Green Version]
- Souza, V.P.; Ikegami, C.M.; Arantes, G.M.; Marana, S.R. Mutations close to a hub residue affect the distant active site of a GH1 β-glucosidase. PLoS ONE 2018, 13, e0198696. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Bandulik, S.; Stindl, J.; Zilbermint, M.; Dedov, I.; Mulatero, P.; Allgaeuer, M.; Lee, C.-C.R.; Stratakis, C.A.; Williams, T.A.; et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J. Clin. Endocrinol. Metab. 2015, 100, E114–E118. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. A neural-network-based method for predicting protein stability changes upon single point mutations. Bioinformatics 2004, 20 (Suppl. 1), i63–i68. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM–A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef]
- Blanco, J.D.; Radusky, L.; Climente-González, H.; Serrano, L. FoldX accurate structural protein-DNA binding prediction using PADA1 (Protein Assisted DNA Assembly 1). Nucleic Acids Res. 2018, 46, 3852–3863. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, L.; Gao, Y.; Zhang, J.; Zhenirovskyy, M.; Alexov, E. Predicting folding free energy changes upon single point mutations. Bioinformatics 2012, 28, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getov, I.; Petukh, M.; Alexov, E. SAAFEC: Predicting the Effect of Single Point Mutations on Protein Folding Free Energy Using a Knowledge-Modified MM/PBSA Approach. Int. J. Mol. Sci. 2016, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Petukh, M.; Alexov, E.; Panchenko, A.R. Predicting the Impact of Missense Mutations on Protein-Protein Binding Affinity. J. Chem. Theory Comput. 2014, 10, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Dai, L.; Alexov, E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. Int. J. Mol. Sci. 2016, 17, 547. [Google Scholar] [CrossRef] [PubMed]
- Cang, Z.; Wei, G.-W. Analysis and prediction of protein folding energy changes upon mutation by element specific persistent homology. Bioinformatics 2017, 33, 3549–3557. [Google Scholar] [CrossRef]
- Cang, Z.; Wei, G.-W. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLOS Comput. Biol. 2017, 13, e1005690. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Shu, W.; Devlin, G.L.; Meehan, S.; Auer, S.; Dobson, C.M.; Welland, M.E. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc. Natl. Acad. Sci. USA 2007, 104, 10016–10021. [Google Scholar] [CrossRef] [Green Version]
- Rivas, G.; Minton, A.P. Macromolecular crowding in vitro, in vivo, and in between. Trends Biochem. Sci. 2016, 41, 970–981. [Google Scholar] [CrossRef]
- Lee, H.-T.; Kilburn, D.; Behrouzi, R.; Briber, R.M.; Woodson, S.A. Molecular crowding overcomes the destabilizing effects of mutations in a bacterial ribozyme. Nucleic Acids Res. 2015, 43, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Senske, M.; Törk, L.; Born, B.; Havenith, M.; Herrmann, C.; Ebbinghaus, S. Protein Stabilization by Macromolecular Crowding through Enthalpy Rather Than Entropy. J. Am. Chem. Soc. 2014, 136, 9036–9041. [Google Scholar] [CrossRef] [PubMed]
- Vreven, T.; Hwang, H.; Pierce, B.G.; Weng, Z. Prediction of protein–protein binding free energies. Protein Sci. 2012, 21, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Domański, J.; Hedger, G.; Best, R.B.; Stansfeld, P.J.; Sansom, M.S.P. Convergence and Sampling in Determining Free Energy Landscapes for Membrane Protein Association. J. Phys. Chem. B 2017, 121, 3364–3375. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, N.M.; Fenley, A.T.; Gilson, M.K. Computational Calorimetry: High-Precision Calculation of Host–Guest Binding Thermodynamics. J. Chem. Theory Comput. 2015, 11, 4377–4394. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Mutations: Types and Causes. Molecular Cell Biology 4th Edition 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK21578/ (accessed on 22 December 2018).
- Gao, M.; Zhou, H.; Skolnick, J. Insights into disease-associated mutations in the human proteome through protein structural analysis. Structure 2015, 23, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Luigi Martelli, P. Correlating disease-related mutations to their effect on protein stability: A large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Michel, J. Current and emerging opportunities for molecular simulations in structure-based drug design. Phys. Chem. Chem. Phys. 2014, 16, 4465–4477. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-L.; Chen, C.-C. Computational approaches for drug discovery. Drug Dev. Res. 2014, 75, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Lounnas, V.; Ritschel, T.; Kelder, J.; McGuire, R.; Bywater, R.P.; Foloppe, N. Current progress in Structure-Based Rational Drug Design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, M.P.; Samara, G.; Hurwitz, M.; Passaro, E. Human Genome Project. Am. J. Surg. 1993, 165, 258–264. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073.
- Peng, Y.; Norris, J.; Schwartz, C.; Alexov, E. Revealing the Effects of Missense Mutations Causing Snyder-Robinson Syndrome on the Stability and Dimerization of Spermine Synthase. Int. J. Mol. Sci. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jia, Z.; Peng, Y.; Godar, S.; Getov, I.; Teng, S.; Alper, J.; Alexov, E. Forces and Disease: Electrostatic force differences caused by mutations in kinesin motor domains can distinguish between disease-causing and non-disease-causing mutations. Sci. Rep. 2017, 7, 8237. [Google Scholar] [CrossRef] [PubMed]
- Spellicy, C.J.; Norris, J.; Bend, R.; Bupp, C.; Mester, P.; Reynolds, T.; Dean, J.; Peng, Y.; Alexov, E.; Schwartz, C.E.; et al. Key apoptotic genes APAF1 and CASP9 implicated in recurrent folate-resistant neural tube defects. Eur. J. Hum. Genet. 2018, 26, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, K.; Niranjan, T.; Selvan, N.; Teo, C.F.; May, M.; Patel, S.; Weatherly, B.; Skinner, C.; Opitz, J.; Carey, J.; et al. Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability. J. Biol. Chem. 2017, 292, 8948–8963. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Hong, C.-J.; Lin, Y.-T.; Chang, W.-H.; Huang, H.-T.; Liao, J.-Y.; Chang, Y.-J.; Hsieh, Y.-F.; Cheng, C.-Y.; Liu, H.-C.; et al. Amyloid-beta (Aβ) D7H mutation increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies. PLoS ONE 2012, 7, e35807. [Google Scholar] [CrossRef] [PubMed]
- Alexov, E. Advances in Human Biology: Combining Genetics and Molecular Biophysics to Pave the Way for Personalized Diagnostics and Medicine. Available online: https://www.hindawi.com/journals/ab/2014/471836/ (accessed on 8 January 2019).
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef]
- Peng, Y.; Myers, R.; Zhang, W.; Alexov, E. Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Peng, Y.; Suryadi, J.; Yang, Y.; Kucukkal, T.G.; Cao, W.; Alexov, E. Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease. Int. J. Mol. Sci. 2015, 16, 27270–27287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Böhm, H.-J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Nair, P.C.; Miners, J.O. Molecular dynamics simulations: From structure function relationships to drug discovery. In Silico Pharmacol 2014, 2, 4. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Wesley Hatfield, G.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Kaar, J.L.; Basse, N.; Joerger, A.C.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Michael, A.J. Spermine synthase. Cell. Mol. Life Sci. 2010, 67, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Martiny, V.; Lagorce, D.; Ikeguchi, Y.; Alexov, E.; Miteva, M.A. Rational Design of Small-Molecule Stabilizers of Spermine Synthase Dimer by Virtual Screening and Free Energy-Based Approach. PLoS ONE 2014, 9, e110884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Witham, S.; Petukh, M.; Moroy, G.; Miteva, M.; Ikeguchi, Y.; Alexov, E. A rational free energy-based approach to understanding and targeting disease-causing missense mutations. J. Am. Med. Inform. Assoc. 2013, 20, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Novel approach for efficient pharmacophore-based virtual screening: Method and applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Huang, H.-C.; Juan, H.-F. Reviewing ligand-based rational drug design: The search for an ATP synthase inhibitor. Int. J. Mol. Sci. 2011, 12, 5304–5318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, H.; Zhang, Q.; Bao, X.; Yu, K.; Luo, X.; Zhu, W.; Jiang, H. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Silakari, O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorg. Chem. 2018, 79, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Springsteel, M.F.; Galietta, L.J.V.; Ma, T.; By, K.; Berger, G.O.; Yang, H.; Dicus, C.W.; Choung, W.; Quan, C.; Shelat, A.A.; et al. Benzoflavone activators of the cystic fibrosis transmembrane conductance regulator: Towards a pharmacophore model for the nucleotide-binding domain. Bioorg. Med. Chem. 2003, 11, 4113–4120. [Google Scholar] [CrossRef]
- Pathak, D.; Chadha, N.; Silakari, O. Identification of non-resistant ROS-1 inhibitors using structure based pharmacophore analysis. J. Mol. Graph. Model. 2016, 70, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, X.; Wu, H.; Zou, F.; Yan, X.-E.; Chen, C.; Hu, C.; Yu, K.; Wang, W.; Zhao, P.; et al. Discovery of (R)-1-(3-(4-Amino-3-(3-chloro-4-(pyridin-2-ylmethoxy)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (CHMFL-EGFR-202) as a Novel Irreversible EGFR Mutant Kinase Inhibitor with a Distinct Binding Mode. J. Med. Chem. 2017, 60, 2944–2962. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740. [Google Scholar] [CrossRef]
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.-L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired Resistance to Crizotinib from a Mutation in CD74–ROS1. N. Engl. J. Med. 2013, 368, 2395–2401. [Google Scholar] [CrossRef] [Green Version]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Noy, E.; Senderowitz, H. Combating cystic fibrosis: In search for CF transmembrane conductance regulator (CFTR) modulators. ChemMedChem 2011, 6, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Liessi, N.; Cichero, E.; Pesce, E.; Arkel, M.; Salis, A.; Tomati, V.; Paccagnella, M.; Damonte, G.; Tasso, B.; Galietta, L.J.V.; et al. Synthesis and biological evaluation of novel thiazole- VX-809 hybrid derivatives as F508del correctors by QSAR-based filtering tools. Eur. J. Med. Chem. 2018, 144, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Konrat, R. The protein meta-structure: A novel concept for chemical and molecular biology. Cell. Mol. Life Sci. 2009, 66, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, Y.; Pons, M.; Konrat, R. Meta-structure correlation in protein space unveils different selection rules for folded and intrinsically disordered proteins. Mol. Biosyst. 2012, 8, 411–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.A.; Waldmann, H. Protein structure similarity clustering and natural product structure as guiding principles in drug discovery. Drug Discov. Today 2005, 10, 471–483. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ascher, D.B.; Thomas, S.E.; Blundell, T.L. Genomes, structural biology and drug discovery: Combating the impacts of mutations in genetic disease and antibiotic resistance. Biochem. Soc. Trans. 2017, 45, 303–311. [Google Scholar] [CrossRef]
- Zhang, Z.; Norris, J.; Schwartz, C.; Alexov, E. In Silico and In Vitro Investigations of the Mutability of Disease-Causing Missense Mutation Sites in Spermine Synthase. PLoS ONE 2011, 6, e20373. [Google Scholar] [CrossRef]
- Frey, K.M.; Georgiev, I.; Donald, B.R.; Anderson, A.C. Predicting resistance mutations using protein design algorithms. Proc. Natl. Acad. Sci. USA 2010, 107, 13707–13712. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, S.; Gilbert, N.; Perry, P.; Östlund, C.; Worman, H.J.; Bickmore, W.A. Altered protein dynamics of disease-associated lamin A mutants. BMC Cell. Biol. 2004, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; De Felice, F.G. PABMB Lecture. Protein dynamics, folding and misfolding: From basic physical chemistry to human conformational diseases. FEBS Lett. 2001, 498, 129–134. [Google Scholar] [CrossRef]
- Cheng, L.S.; Amaro, R.E.; Xu, D.; Li, W.W.; Arzberger, P.W.; McCammon, J.A. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J. Med. Chem. 2008, 51, 3878–3894. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, C.; Michel, H. Crystallization of membrane proteins. Curr. Opin. Struct. Biol. 1997, 7, 697–701. [Google Scholar] [CrossRef]
- Lluis, M.W.; Godfroy, J.I.; Yin, H. Protein engineering methods applied to membrane protein targets. Protein Eng. Des. Sel. 2013, 26, 91–100. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aided Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Niu, X.; Jin, F.; Liu, Z.; Jin, C.; Lai, L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016, 6, 22298. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.-C.; Tou, W.I. How to design a drug for the disordered proteins? Drug Discov. Today 2013, 18, 910–915. [Google Scholar] [CrossRef]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [Green Version]
- Perez-Aguilar, J.M.; Saven, J.G. Computational Design of Membrane Proteins. Structure 2012, 20, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Alford, R.F.; Koehler Leman, J.; Weitzner, B.D.; Duran, A.M.; Tilley, D.C.; Elazar, A.; Gray, J.J. An Integrated Framework Advancing Membrane Protein Modeling and Design. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Patra, H.K.; Islam, M.; Basu, S.; Griffith, M. Peptide Architectonics for Biotherapeutics. Indian Patent Application No. 201741036721; Filed on 16 October 2017,





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Alexov, E.; Basu, S. Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. Int. J. Mol. Sci. 2019, 20, 548. https://doi.org/10.3390/ijms20030548
Peng Y, Alexov E, Basu S. Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. International Journal of Molecular Sciences. 2019; 20(3):548. https://doi.org/10.3390/ijms20030548
Chicago/Turabian StylePeng, Yunhui, Emil Alexov, and Sankar Basu. 2019. "Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases" International Journal of Molecular Sciences 20, no. 3: 548. https://doi.org/10.3390/ijms20030548
APA StylePeng, Y., Alexov, E., & Basu, S. (2019). Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. International Journal of Molecular Sciences, 20(3), 548. https://doi.org/10.3390/ijms20030548