Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity



Abstract
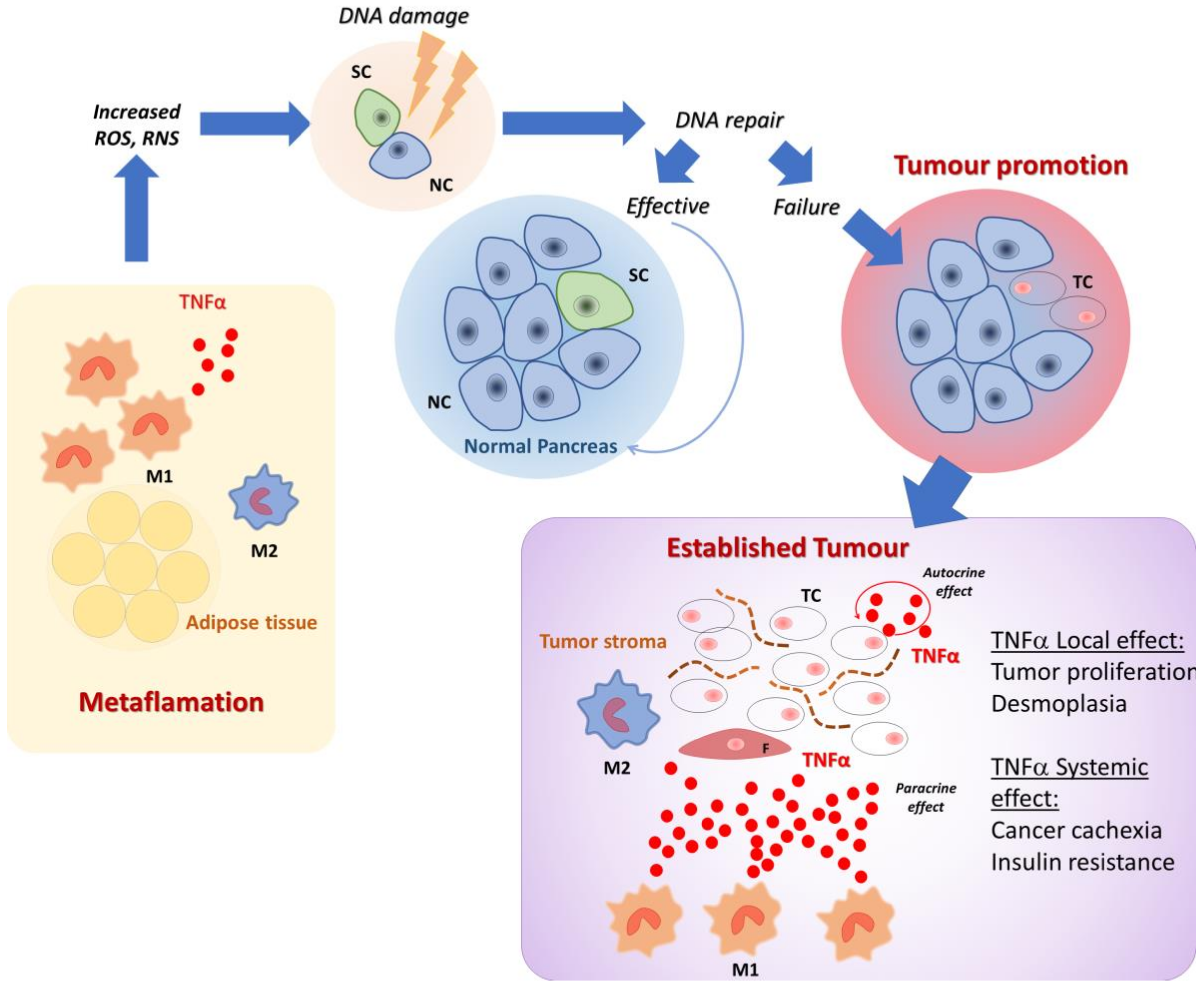
:1. Introduction
2. Role of Inflammation, Diabetes, and Obesity in Enhancing PDAC Risk
3. The Role of Local Inflammation in PDAC Growth, Development, and Metastasis
4. Cytokines Released by Immune and Tumor Cells, and Their Role in Inflammatory Response to Cancer Cells
5. Cytokines Are Involved in Cachexia and Cancer Induced Metabolic Alterations
6. Immune Response to Cancer Cells
6.1. Myeloid-Derived Suppressor Cells
6.2. Tumour Associated Macrophages (TAM)
6.3. Treg Cells
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, pancreatogenic diabetes, and pancreatic cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef]
- Bosetti, C.; Rosato, V.; Li, D.; Silverman, D.; Petersen, G.M.; Bracci, P.M.; Neale, R.E.; Muscat, J.; Anderson, K.; Gallinger, S.; et al. Diabetes, antidiabetic medications, and pancreatic cancer risk: an analysis from the International Pancreatic Cancer Case-Control Consortium. Ann. Oncol. 2014, 25, 2065–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkegàrd, J.; Mortensen, F.V.; Cronin-Fenton, D. Chronic pancreatitis and pancreatic cancer risk: a systematic review and meta-analysis. Am. J. Gastroenterol. 2017, 112, 1366–1372. [Google Scholar] [CrossRef]
- Pang, Y.; Kartsonaki, C.; Guo, Y.; Bragg, F.; Yang, L.; Bian, Z.; Chen, Y.; Iona, A.; Millwood, I.Y.; Lv, J.; et al. Diabetes, plasma glucose and incidence of pancreatic cancer: A prospective study of 0.5 million Chinese adults and a meta-analysis of 22 cohort studies. Int. J. Cancer 2017, 140, 1781–1788. [Google Scholar] [CrossRef] [Green Version]
- Koyanagi, Y.N.; Matsuo, K.; Ito, H.; Tamakoshi, A.; Sugawara, Y.; Hidaka, A.; Wada, K.; Oze, I.; Kitamura, Y.; Liu, R.; et al. Body-mass index and pancreatic cancer incidence: a pooled analysis of nine population-based cohort studies with more than 340,000 Japanese subjects. J. Epidemiol. 2018, 28, 245–252. [Google Scholar] [CrossRef]
- Rothwell, PM.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Arem, H.; Reedy, J.; Sampson, J.; Jiao, L.; Hollenbeck, A.R.; Risch, H.; Mayne, S.T.; Stolzenberg-Solomon, R.Z. The healthy eating index 2005 and risk for pancreatic cancer in the NIH-AARP study. J. Natl. Cancer Inst. 2013, 105, 1298–1305. [Google Scholar] [CrossRef]
- Risch, H.A.; Lu, L.; Streicher, S.A.; Wang, J.; Zhang, W.; Ni, Q.; Kidd, M.S.; Yu, H.; Gao, Y.T. Aspirin use and reduced risk of pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Yellow, W.; Bamlet, W.R.; Oberg, A.L.; Anderson, K.E.; Olson, J.E.; Sinha, R.; Petersen, G.M.; Stolzenberg-Solomon, R.Z.; Jansen, R.J. Association between alcohol consumption, folate intake, and risk of pancreatic cancer: a case-control study. Nutrients 2017, 9, E0448. [Google Scholar] [PubMed]
- Jiao, L.; Chen, L.; White, D.L.; Tinker, L.; Chlebowski, R.T.; Van Horn, L.V.; Richardson, P.; Lane, D.; Sangi-Haghpeykar, H.; El-Serag, HB. Low-fat dietary pattern and pancreatic cancer risk in the women's health initiative dietary modification randomized controlled trial. J. Natl. Cancer Inst. 2018, 110, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Virchow, R. Die Krankhaften Geschwülste; Springer: Berlin, Germany, 1863. [Google Scholar]
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Zambon, C.F.; Letley, D.P.; Stranges, A.; Marchet, A.; Rhead, J.L.; Schiavon, S.; Guariso, G.; Ceroti, M.; Nitti, D.; et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 2008, 135, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Wei, C.; Ensor, J.E.; Smolenski, D.J.; Amos, C.I.; Levin, B.; Berry, D.A. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control. 2013, 24, 1207–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatenby, P.; Caygill, C.; Wall, C.; Bhatacharjee, S.; Ramus, J.; Watson, A.; Winslet, M. Lifetime risk of esophageal adenocarcinoma in patients with Barrett's esophagus. World J. Gastroenterol. 2014, 20, 9611–9617. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Everhart, J.; Wright, D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA 1995, 273, 1605–1609. [Google Scholar] [CrossRef]
- Berrington de Gonzalez, A.; Sweetland, S.; Spencer, E. A meta-analysis of obesity and the risk of pancreatic cancer. Br. J. Cancer 2003, 89, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de González, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.J.; Roddam, A.W.; Beral, V. Pancreatic cancer in type 1 and young-onset diabetes: systematic review and meta-analysis. Br. J. Cancer 2007, 96, 507–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Tang, H.; Hassan, M.M.; Holly, E.A.; Bracci, P.M.; Silverman, D.T. Diabetes and risk of pancreatic cancer: a pooled analysis of three large case-control studies. Cancer Causes Control. 2011, 22, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Elena, J.W.; Steplowski, E.; Yu, K.; Hartge, P.; Tobias, G.S.; Brotzman, M.J.; Chanock, S.J.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; et al. Diabetes and risk of pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium. Cancer Causes Control. 2013, 24, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Batabyal, P.; Vander Hoorn, S.; Christophi, C.; Nikfarjam, M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann. Surg. Oncol. 2014, 21, 2453–2462. [Google Scholar] [CrossRef]
- Pannala, R.; Leirness, J.B.; Bamlet, W.R.; Basu, A.; Petersen, G.M.; Chari, S.T. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008, 134, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Moz, S.; Basso, D.; Padoan, A.; Bozzato, D.; Fogar, P.; Zambon, C.F.; Pelloso, M.; Sperti, C.; Vigili de Kreutzenberg, S.; Pasquali, C.; et al. Blood expression of matrix metalloproteinases 8 and 9 and of their inducers S100A8 and S100A9 supports diagnosis and prognosis of PDAC-associated diabetes mellitus. Clin. Chim Acta. 2016, 456, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Marino, A.; Consonni, F.M.; Bleve, A.; Mola, S.; Storto, M.; Riboldi, E.; Sica, A. Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinogenesis 2018, 39, 1095–1104. [Google Scholar] [CrossRef]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef]
- Basso, D.; Gnatta, E.; Padoan, A.; Fogar, P.; Furlanello, S.; Aita, A.; Bozzato, D.; Zambon, C.F.; Arrigoni, G.; Frasson, C.; et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget 2017, 8, 84928–84944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Diet boosts the effectiveness of a cancer drug. Nature 2018, 560, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. The immune revolution: a case for priming, not checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Jordan, B.F. Gut microbiota-mediated inflammation in obesity: a link with gastrointestinal cancer. Nat. Rev. Gastroenterol Hepatol. 2018, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes. 2012, 3, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhou, H.; Zhang, A.; Gao, R.; Yang, S.; Zhao, C.; Wang, Y.; Hu, J.; Goswami, R.; Gong, L.; et al. Serum LBP is associated with insulin resistance in women with PCOS. PLoS One 2016, 11, e0145337. [Google Scholar]
- Awoyemi, A.; Trøseid, M.; Arnesen, H.; Solheim, S.; Seljeflot, I. Markers of metabolic endotoxemia as related to metabolic syndrome in an elderly male population at high cardiovascular risk: a cross-sectional study. Diabetol. Metab. Syndr. 2018, 10, 59. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient competition: a new axis of tumor immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.; Costello, E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 2012, 3, 270. [Google Scholar] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Inman, K.S.; Francis, A.A.; Murray, N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11160–11181. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Potenza, A.; Andriulli, A.; Pazienza, V. Exploring the microbiota to better understand gastrointestinal cancers physiology. Clin. Chem Lab. Med. 2018, 56, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Adamberg, K.; Jaagura, M.; Copetti, M.; Fontana, A.; Adamberg, S.; Kolk, K.; Vilu, R.; Andriulli, A.; Pazienza, V. Influence of gemcitabine chemotherapy on the microbiota of pancreatic cancer xenografted mice. Cancer Chemother. Pharmacol. 2018, 81, 773–782. [Google Scholar] [CrossRef]
- Basso, D.; Bozzato, D.; Padoan, A.; Moz, S.; Zambon, C.F.; Fogar, P.; Greco, E.; Scorzeto, M.; Simonato, F.; Navaglia, F.; et al. Inflammation and pancreatic cancer: molecular and functional interactions between S100A8, S100A9, NT-S100A8 and TGFβ1. Cell Commun. Signal. 2014, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.; Combs, S.E.; Springfeld, C.; Hartwig, W.; Hackert, T.; Büchler, M.W. Advanced-stage pancreatic cancer: therapy options. Nat. Rev. Clin. Oncol. 2013, 10, 323–333. [Google Scholar] [CrossRef]
- Chandrasegaram, M.D.; Goldstein, D.; Simes, J.; Gebski, V.; Kench, J.G.; Gill, A.J.; Samra, J.S.; Merrett, N.D.; Richardson, A.J.; Barbour, A.P. Meta-analysis of radical resection rates and margin assessment in pancreatic cancer. Br. J. Surg. 2015, 102, 1459–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.A.; Vimalachandran, D.; Thompson, C.C.; Jenkins, R.E.; Nedjadi, T.; Shekouh, A.; Campbell, F.; Dodson, A.; Prime, W.; Crnogorac-Jurcevic, T.; et al. The expression of S100A8 in pancreatic cancer-associated monocytes is associated with the Smad4 status of pancreatic cancer cells. Proteomics 2007, 7, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.W.; Nedjadi, T.; Sheikh, A.A.; Tweedle, E.M.; Tonack, S.; Honap, S.; Jenkins, R.E.; Park, B.K.; Schwarte-Waldhoff, I.; Khattak, I.; et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis 2010, 31, 1541–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Tumor-infiltrating monocytes/macrophages promote tumor invasion and migration by upregulating S100A8 and S100A9 expression in cancer cells. Oncogene 2016, 35, 5735–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenblaetter, M.; Flores-Borja, F.; Lee, J.J.; Wefers, C.; Smith, H.; Hueting, R.; Cooper, M.S.; Blower, P.J.; Patel, D.; Rodriguez-Justo, M.; et al. Visualization of tumor-immune interaction - Target-specific imaging of S100A8/A9 reveals pre-metastatic niche establishment. Theranostics 2017, 7, 2392–2401. [Google Scholar] [CrossRef] [PubMed]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Yako, Y.Y.; Kruger, D.; Smith, M.; Brand, M. Cytokines as biomarkers of pancreatic ductal adenocarcinoma: a systematic review. PLoS One 2016, 11, e0154016. [Google Scholar] [CrossRef]
- Karakhanova, S.; Link, J.; Heinrich, M.; Shevchenko, I.; Yang, Y.; Hassenpflug, M.; Bunge, H.; von Ahn, K.; Brecht, R.; Mathes, A.; et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: importance of myeloid-derived suppressor cells. Oncoimmunology 2015, 4, e998519. [Google Scholar] [CrossRef] [Green Version]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Krüger, U.; Becker, T.; Ebsen, M.; Röcken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861. [Google Scholar] [CrossRef] [Green Version]
- Pop, V.V.; Seicean, A.; Lupan, I.; Samasca, G.; Burz, C.C. IL-6 roles - Molecular pathway and clinical implication in pancreatic cancer - A systemic review. Immunol. Lett. 2017, 181, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nab-paclitaxel interrupts cancer-stromal interaction through C-X-C motif chemokine 10-mediated interleukin-6 downregulation in vitro. Cancer Sci. 2018, 109, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Basso, D.; Fogar, P.; Mazza, S.; Navaglia, F.; Zambon, C.F.; Falda, A.; Pedrazzoli, S.; Ancona, E.; Plebani, M. Pancreatic cancer cells invasiveness is mainly affected by interleukin-1beta not by transforming growth factor-beta1. Int. J. Biol. Markers 2005, 20, 235–241. [Google Scholar] [CrossRef]
- Nomura, A.; Gupta, V.K.; Dauer, P.; Sharma, N.S.; Dudeja, V.; Merchant, N.; Saluja, A.K.; Banerjee, S. NFkB-mediated invasiveness in CD133(+) pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol. Cancer Res. 2018, 16, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. Tumor-promoting inflammatory networks in pancreatic neoplasia: Another reason to loathe Kras. Cancer Cell 2014, 25, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Loncle, C.; Bonjoch, L.; Folch-Puy, E.; Lopez-Millan, M.B.; Lac, S.; Molejon, M.I.; Chuluyan, E.; Cordelier, P.; Dubus, P.; Lomberk, G.; et al. IL17 functions through the novel REG3b-JAK2-STAT3 inflammatory pathway to promote the transition from chronic pancreatitis to pancreatic cancer. Cancer Res. 2015, 75, 4852–4862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zoltan, M.; Riquelme, E.; Xu, H.; Sahin, I.; Castro-Pando, S.; Montiel, M.F.; Chang, K.; Jiang, Z.; Ling, J.; et al. Immune cell production of Interleukin 17 induces stem cell features of pancreatic intraepithelial neoplasia cells. Gastroenterology 2018, 155, 210–223. [Google Scholar] [CrossRef]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van de Velde, L.A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Araki, K.; Yokobori, T.; Hagiwara, K.; Gantumur, D.; Yamanaka, T.; Handa, T.; Tsukagoshi, M.; Igarashi, T.; Watanabe, A.; et al. Conophylline suppresses pancreatic cancer desmoplasia and cancer-promoting cytokines produced by cancer-associated fibroblasts. Cancer Sci. 2019, 110, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Perales, S.; Alejandre, M.J.; Iglesias, J.; Palomino, R.J.; Martin, M.; Caba, O.; Prados, J.C.; Aránega, A.; Delgado, J.R.; et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas 2014, 43, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A new venue of TNF targeting. Int J. Mol. Sci. 2018, 19, E1442. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, G.; Li, Q.; Wang, Z.; Hu, W.; Li, P.; Li, S.; Wu, H.; Kong, X.; Gao, J.; et al. Hedgehog signaling non-canonical activated by pro-inflammatory cytokines in pancreatic ductal adenocarcinoma. J. Cancer 2016, 7, 2067–2076. [Google Scholar] [CrossRef]
- Chopra, M.; Lang, I.; Salzmann, S.; Pachel, C.; Kraus, S.; Bäuerlein, C.A.; Brede, C.; Garrote, A.L.; Mattenheimer, K.; Ritz, M.; et al. Tumor necrosis factor induces tumor promoting and anti-tumoral effects on pancreatic cancer via TNFR1. PLoS One 2013, 8, e75737. [Google Scholar] [CrossRef] [PubMed]
- Kali, A.; Ostapchuk, Y.O.; Belyaev, N.N. TNFα and TGFβ-1 synergistically increase the cancer stem cell properties of MiaPaCa-2 cells. Oncol. Lett. 2017, 14, 4647–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, J.M.; Wild, A.T.; Wang, H.; Tran, P.T.; Chang, K.J.; Taylor, G.E.; Donehower, R.C.; Pawlik, T.M.; Ziegler, M.A.; Cai, H.; et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: final results. J. Clin. Oncol. 2013, 31, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Fernandez, S.A.; Criswell, T.; Chidiac, T.A.; Guttridge, D.; Villalona-Calero, M.; Bekaii-Saab, T.S. Disrupting cytokine signaling in pancreatic cancer: a phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 2013, 42, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Fahrbach, K.; Nordstrom, B.; Ross, S.; Schmid, C.H.; Symmons, D. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011, 20, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Olivo, M.A.; Tayar, J.H.; Martinez-Lopez, J.A.; Pollono, E.N.; Cueto, J.P.; Gonzales-Crespo, M.R.; Fulton, S.; Suarez-Almazor, M.E. Risk of malignancies in patients with rheumatoid arthritis treated with biologic therapy: a meta-analysis. JAMA 2012, 308, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Peyrin-Biroulet, L.; Ford, A.C. Systematic review with meta-analysis: malignancies with anti-tumour necrosis factor-α therapy in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2014, 39, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Lara, S.; Johnson, J.; Kulkarni, P. New pancreatic adenocarcinoma in a Crohn's patient treated with tumor necrosis factor (TNF) inhibitors for 6 months. J. Gastrointest. Cancer 2014, 45, Suppl 1. 226–229. [Google Scholar] [CrossRef]
- Hawa, Z.; Haque, I.; Ghosh, A.; Banerjee, S.; Harris, L.; Banerjee, S.K. The miRacle in Pancreatic Cancer by miRNAs: Tiny Angels or Devils in Disease Progression. Int. J. Mol. Sci. 2016, 17, 809. [Google Scholar] [CrossRef]
- dbDEMC 2.0, A Database of Differentially Expressed miRNAs in Human Cancers (version 2.0). Available online: http://www.picb.ac.cn/dbDEMC/ (accessed on 6 December 2018).
- Ottaviani, S.; Stebbing, J.; Frampton, A.E.; Zagorac, S.; Krell, J.; de Giorgio, A.; Trabulo, S.M.; Nguyen, V.T.M.; Magnani, L.; Feng, H.; et al. TGF-β induces miR-100 and miR-125b but blocks let-7a through LIN28B controlling PDAC progression. Nat. Commun. 2018, 9, 1845. [Google Scholar] [CrossRef] [Green Version]
- Moz, S.; Basso, D.; Bozzato, D.; Galozzi, P.; Navaglia, F.; Negm, O.H.; Arrigoni, G.; Zambon, C.F.; Padoan, A.; Tighe, P.; et al. SMAD4 loss enables EGF, TGFβ1 and S100A8/A9 induced activation of critical pathways to invasion in human pancreatic adenocarcinoma cells. Oncotarget 2016, 7, 69927–69944. [Google Scholar] [CrossRef] [Green Version]
- Papaconstantinou, I.; Kapizioni, C.; Legaki, E.; Xourgia, E.; Karamanolis, G.; Gklavas, A.; Gazouli, M. Association of miR-146 rs2910164, miR-196a rs11614913, miR-221 rs113054794 and miR-224 rs188519172 polymorphisms with anti-TNF treatment response in a Greek population with Crohn's disease. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Pivarcsi, A.; Meisgen, F.; Xu, N.; Ståhle, M.; Sonkoly, E. Changes in the level of serum microRNAs in patients with psoriasis after antitumour necrosis factor-α therapy. Br. J. Dermatol. 2013, 169, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, M.; Bonek, K.; Merdas, M.; Zarecki, P.; Swierkot, J.; Gluszko, P.; Bogunia-Kubik, K.; Maslinski, W. Changes in MiRNA-5196 expression as a potential biomarker of anti-TNF-α therapy in rheumatoid arthritis and ankylosing spondylitis patients. Arch. Immunol Ther Exp. (Warsz) 2018, 66, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Castro-Villegas, C.; Pérez-Sánchez, C.; Escudero, A.; Filipescu, I.; Verdu, M.; Ruiz-Limón, P.; Aguirre, M.A.; Jiménez-Gomez, Y.; Font, P.; Rodriguez-Ariza, A.; et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Arthritis. Res. Ther. 2015, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Krintel, S.B.; Dehlendorff, C.; Hetland, M.L.; Hørslev-Petersen, K.; Andersen, K.K.; Junker, P.; Pødenphant, J.; Ellingsen, T.; Ahlquist, P.; Lindegaard, H.M.; et al. Prediction of treatment response to adalimumab: a double-blind placebo-controlled study of circulating microRNA in patients with early rheumatoid arthritis. Pharmacogenomics J. 2016, 16, 141–146. [Google Scholar]
- Fujioka, S.; Nakamichi, I.; Esaki, M.; Asano, K.; Matsumoto, T.; Kitazono, T. Serum microRNA levels in patients with Crohn's disease during induction therapy by infliximab. J. Gastroenterol. Hepatol. 2014, 29, 1207–1214. [Google Scholar] [CrossRef]
- Cuppen, B.V.; Rossato, M.; Fritsch-Stork, R.D.; Concepcion, A.N.; Schenk, Y.; Bijlsma, J.W.; Radstake, T.R.; Lafeber, F.P.; on behalf of all SRU investigators. Can baseline serum microRNAs predict response to TNF-alpha inhibitors in rheumatoid arthritis? Arthritis Res. Ther. 2016, 18, 189. [Google Scholar] [CrossRef] [Green Version]
- miRBase: the microRNA database. Available online: http://mirbase.org/help/ (accessed on 6 December 2018).
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 11, e493–e503. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [Green Version]
- Patel, H.J.; Patel, B.M. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Greco, E.; Padoan, A.; Fogar, P.; Scorzeto, M.; Fadi, E.; Bozzato, D.; Moz, S.; Navaglia, F.; Zambon, C.F.; et al. Altered intracellular calcium fluxes in pancreatic cancer induced diabetes mellitus: Relevance of the S100A8 N-terminal peptide (NT-S100A8). J. Cell Physiol. 2011, 226, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; van Eijck, C.H.; Peppelenbosch, M.P.; Sleijfer, S.; Bruno, M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat. Rev. 2014, 40, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.S.; Lee, H.M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Fogar, P.; Falconi, M.; Fadi, E.; Sperti, C.; Frasson, C.; Greco, E.; Tamburrino, D.; Teolato, S.; Moz, S.; et al. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PLoS One 2013, 8, e54824. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Hu, J.; Wang, M.; Peng, F.; Tian, R.; Guo, X.J.; Xie, Y.; Qin, R.Y. Circulating myeloid-derived suppressor cells in patients with pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2016, 15, 99–105. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 2018, 28, 87–103. [Google Scholar] [CrossRef]
- Ratnam, N.M.; Peterson, J.M.; Talbert, E.E.; Ladner, K.J.; Rajasekera, P.V.; Schmidt, C.R.; Dillhoff, M.E.; Swanson, B.J.; Haverick, E.; Kladney, R.D.; et al. NF-κB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J. Clin. Invest. 2017, 127, 3796–3809. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Grage-Griebenow, E.; Schäfer, H.; Sebens, S. The fatal alliance of cancer and T cells: How pancreatic tumor cells gather immunosuppressive T cells. Oncoimmunology 2014, 3, e29382. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.J.; Ma, J.Y.; Hu, G. Lymphocyte-to-monocyte ratio in pancreatic cancer: Prognostic significance and meta-analysis. Clin. Chim. Acta. 2018, 481, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tao, L.; Lu, M.; Xiu, D. Prognostic role of platelet to lymphocyte ratio in pancreatic cancers: A meta-analysis including 3028 patients. Medicine (Baltimore) 2018, 97, e9616. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wei, Q.; Fan, J.; Cheng, S.; Ding, W.; Hua, Z. Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: A meta-analysis containing 8252 patients. Clin. Chim. Acta. 2018, 479, 181–189. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Pro-Inflammatory Cytokines | ||||
| Cytokine | Cell sources | PDAC cells production in vitro | Effects on immune cells | Effects on PDAC initiation and EMT |
| TGF [58,60,61,62,63] | M2 macrophages, Th2 lymphocytes, and TAM | Yes—high | Promotes immune evasion and tolerogenic DC | Inhibits cell cycle progression in early stages, enhances invasion and metastasis by inducing EMT in advanced stages |
| IL-10 [60,61] | M2 macrophages, Treg, Mast cells, and TAM | Yes—high | Promotes immune evasion | PDAC associated TAM have a mixed M1 and M2 phenotype, produce high amounts of IL-10, IL-1β, IL-6 and TNFα and induce EMT in early tumorigenesis |
| Pro-Inflammatory Cytokines | ||||
| Cytokine | Cell sources | PDAC cells production in vitro | Effects on immune cells | Effects on PDAC initiation and EMT |
| IL-6 [58,60,62,63] | CAFs and TAM | Yes—high | Promotes Th2 type cytokine production | Promotes oncogenesis through JAK2-STAT3 activation, angiogenesis through the induction of VEGF, cancer cell migration and EMT |
| IL-1β [58,60,61,64,65] | DC, M1 macrophages, and TAM | Yes—low | Recruitment of MDSC and T-cell activation by inducing the production of IL-2 and IL-2R | Promotes cancer growth, invasion, and metastases |
| IL-17 [60,66,67,68,69] | Th 17 CD4+ cells | Yes—low | Recruitment of MDSC | Induces stemness, tumor initiation, and progression, not complete EMT. The expression of the IL-17 receptor is evident on cancer cells undergoing EMT, and depends on oncogenic Kras |
| TNFα [46,60,61,70,71,72] | M1 macrophages, TAM, neutrophils, mast cells, and pancreatic stellate cells | Yes—low | Antagonizes M2 macrophages polarization | Associated with PDAC initiation. Promotes angiogenesis by inducing VEGF production by fibroblasts and metastases by activating NF-β signaling |
| miRNA | Up or Down-Regulated in PDAC [91] * | Average logFold Change * | Up or Down-Regulated by Anti TNFα Therapy | Anti-TNFα Therapy |
|---|---|---|---|---|
| hsa-miR-146 # polymorphism | Found downregulated in one study * | - | No correlation | Crohn’s disease treated with infliximab or adalimumab [94]. |
| hsa-miR-196a # polymorphism | Found upregulated in three studies * | No correlation | ||
| hsa-miR-221 polymorphism | Found upregulated in five studies | 1.9 | No correlation | |
| hsa-miR-224 polymorphism | Found upregulated in four studies | 1.8 | No correlation | |
| hsa-miR-106b | Found upregulated in seven studies cancer/normal | 1.33 | Downregulated in treated patients | Psoriasis treated with etanercept [95]. |
| hsa-miR-26b | Found upregulated in one study/downregulated in three studies | −0.9 | Downregulated in treated patients | |
| hsa-miR-143-3p | - | Downregulated in treated patients | ||
| hsa-miR-223 | Found upregulated in three studies/downregulated in one study | 1.11 | Downregulated in treated patients | |
| hsa-miR-126 | Found upregulated in one study/downregulated in one study | −2.41 | Downregulated in treated patients | |
| hsa-miR-5196 | Not found | - | Downregulated in treated patients | Rheumatoid arthritis and ankylosing spondylitis treated with anti-TNFα treatment (Golimumab in 15%, Adalimumab in 77%, Certolizumab in 8%) [96]. |
| hsa-miR-125b # | Found upregulated in two studies/downregulated in three studies * | −0.28 | Upregulated in treated patients | Rheumatoid arthritis treated with infliximab or etanercept or adalimumab [97]. |
| hsa-miR-126-3p | Found downregulated in one study | −0.70 | Upregulated in treated patients | |
| hsa-miR-146a-5p | Found downregulated in one study | −0.22 | Upregulated in treated patients (see above) | |
| hsa-miR-16-5p | Found downregulated in one study | −0.92 | Upregulated in treated patients | |
| hsa-miR-23-3p | Not found | - | ||
| hsa-miR-223-3p | Found downregulated in one study | −1.17 | Upregulated in treated patients | |
| hsa-miR-22 | Found upregulated in one study | 1.23 | Downregulated in Adalimumab respondent patients | Rheumatoid arthritis treated with Adalimumab [98]. |
| hsa-miR-886-3p | - | - | Upregulated in Adalimumab respondent patients | |
| hsa-let-7d | Found upregulated in 3 studies/Downregulated in 1 study | 0.54 | Upregulated in treated patients | Crohn’s disease treated with infliximab [99]. |
| hsa-let-7e | Found upregulated in 1 studies/Downregulated in 1 study | 0.33 | Upregulated in treated patients | |
| hsa-miR-28-5p | Found upregulated in 2 studies/Downregulated in 1 study | 0.64 | Upregulated in treated patients | |
| hsa-miR-221 | Found upregulated in 5 studies | 1.9 | Upregulated in treated patients (see above) | |
| hsa-miR-224 | Found upregulated in 4 studies | 1.8 | Upregulated in treated patients (see above) | |
| hsa-miR-99a | Found upregulated in 2studies/Downregulated in 3 studies | −0.60 | Upregulated in Adalimumab respondent | Rheumatoid arthritis treated with adalimumab or etanercept [100]. |
| hsa-miR-143 | Found upregulated in 3 studies/Downregulated in 2 studies | −1.05 | Downregulated in respondent | |
| hsa-miR-23a | Found upregulated in 6 studies | 1.29 | Upregulated in respondent | |
| hsa-miR-197 | Found upregulated in 5 studies/Downregulated in 1 study | 1.20 | Upregulated in respondent |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. https://doi.org/10.3390/ijms20030676
Padoan A, Plebani M, Basso D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. International Journal of Molecular Sciences. 2019; 20(3):676. https://doi.org/10.3390/ijms20030676
Chicago/Turabian StylePadoan, Andrea, Mario Plebani, and Daniela Basso. 2019. "Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity" International Journal of Molecular Sciences 20, no. 3: 676. https://doi.org/10.3390/ijms20030676
APA StylePadoan, A., Plebani, M., & Basso, D. (2019). Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. International Journal of Molecular Sciences, 20(3), 676. https://doi.org/10.3390/ijms20030676