Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles

 ,
,  ,
,  , ,
, ,  ,
, 
{kind=link}
Abstract
:1. Introduction
2. Biogenesis and Characterization of Extracellular Vesicles
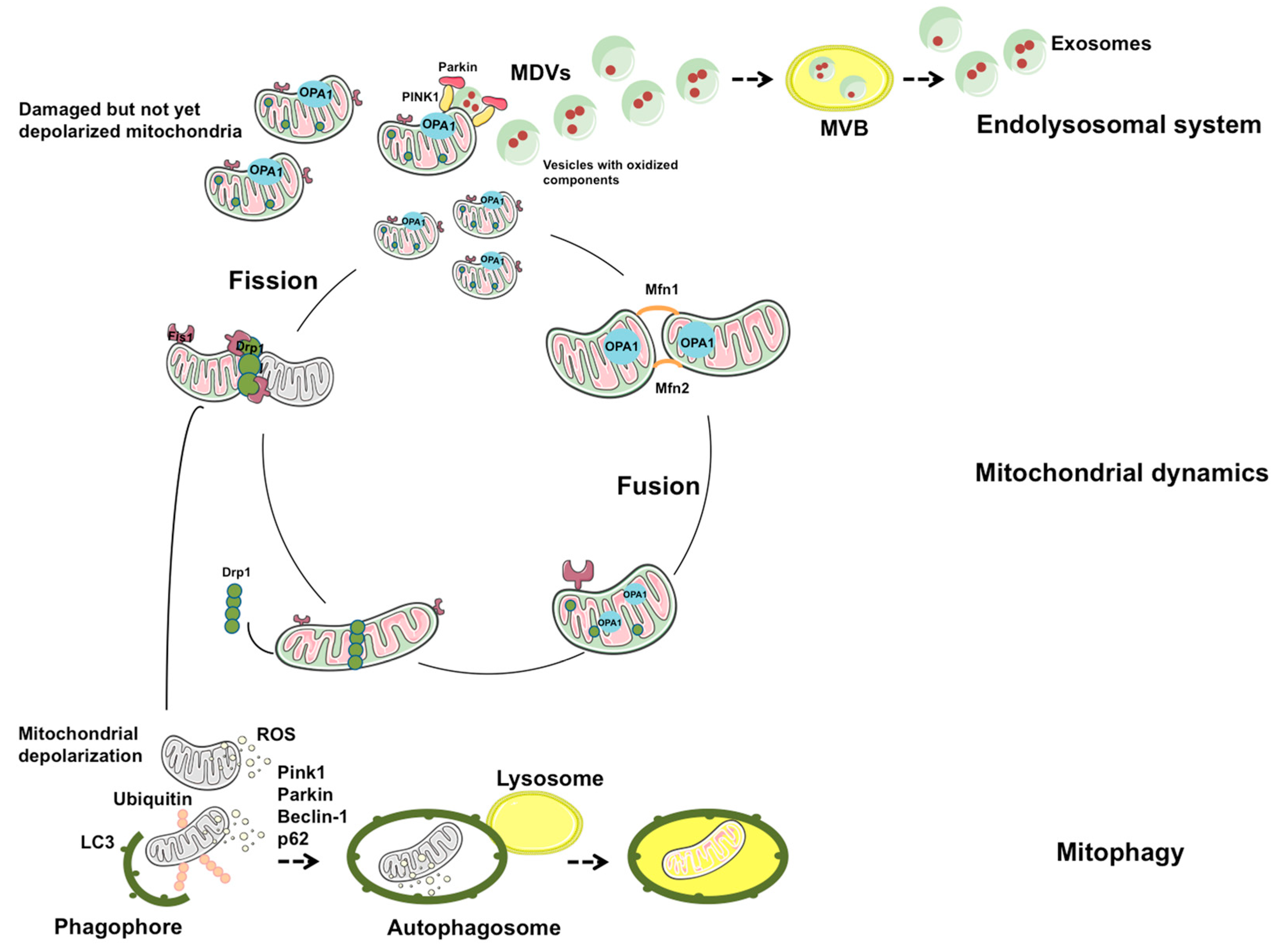
3. The Emerging Role of the Mitochondrial–Lysosomal Axis in Mitochondrial Quality Control
4. Extracellular Vesicle Trafficking and Aging
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AβOs | Amyloid-β oligomers |
| AIF | Apoptosis inducing factor |
| Atg | Autophagy-related gene |
| DRP1 | Dynamin-related protein 1 |
| ESCRT | Endosomal sorting complex required for transport |
| EV | Extracellular vesicle |
| FIS1 | Mitochondrial fission 1 protein |
| ILV | Intraluminal vesicle |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| MDV | Mitochondrial-derived vesicle |
| MFN | Mitofusin |
| MQC | Mitochondrial quality control |
| mtDNA | Mitochondrial DNA |
| mTORC1 | Lysosomal mechanistic target of rapamycin complex 1 |
| MVBs | Multivesicular bodies |
| NiMA | Nutrient-induced Mitochondrial Activity |
| NLRP3 | Nucleotide-binding and leucine-rich repeat receptor (NLR) family pyrin domain containing 3 |
| OPA1 | Optic atrophy-1 |
| PINK1 | Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 |
| RAB | Ras-related protein in Brain |
| RAL1 | Ras Like-1 |
| ROS | Reactive oxygen species |
| SASP | Senescence-associated secretory phenotype |
| TFAM | Mitochondrial transcription factor A |
References
- Landi, F.; Calvani, R.; Cesari, M.; Tosato, M.; Maria Martone, A.; Ortolani, E.; Savera, G.; Salini, S.; Sisto, A.; Picca, A.; et al. Sarcopenia: an overview on current definitions, diagnosis and treatment. Curr. Protein Pept. Sci. 2018, 19, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Marzetti, E.; Thiem, U.; Pérez-Zepeda, M.U.; Abellan Van Kan, G.; Landi, F.; Petrovic, M.; Cherubini, A.; Bernabei, R. The geriatric management of frailty as paradigm of “The end of the disease era”. Eur. J. Intern. Med. 2016, 31, 11–14. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a tool to target aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Burch, J.B.; Augustine, A.D.; Frieden, L.A.; Hadley, E.; Howcroft, T.K.; Johnson, R.; Khalsa, P.S.; Kohanski, R.A.; Li, X.L.; Macchiarini, F.; et al. Advances in geroscience: impact on healthspan and chronic disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S1–S3. [Google Scholar] [CrossRef]
- Espeland, M.A.; Crimmins, E.M.; Grossardt, B.R.; Crandall, J.P.; Gelfond, J.A.L.; Harris, T.B.; Kritchevsky, S.B.; Manson, J.E.; Robinson, J.G.; Rocca, W.A.; et al. Clinical trials targeting aging and age-related multimorbidity. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: linking aging to chronic disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to slow aging in humans: are we ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef]
- Sierra, F. The emergence of geroscience as an interdisciplinary approach to the enhancement of health span and life span. Cold Spring Harb. Perspect. Med. 2016, 6, a025163. [Google Scholar] [CrossRef] [PubMed]
- Schifferli, J.A. Microvesicles are messengers. Semin. Immunopathol. 2011, 33, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; Weber, C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ. Res. 2010, 107, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Kim, O.Y.; Gho, Y.S. Extracellular vesicles as emerging intercellular communicasomes. BMB Rep. 2014, 47, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.M.; Luzio, J.P. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. The sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem. J. 1991, 274, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [PubMed]
- Mitchell, P.; Petfalski, E.; Shevchenko, A.; Mann, M.; Tollervey, D. The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′–>5′ exoribonucleases. Cell 1997, 91, 457–466. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyenne, V.; Apaydin, A.; Rodriguez, D.; Spiegelhalter, C.; Hoff-Yoessle, S.; Diem, M.; Tak, S.; Lefebvre, O.; Schwab, Y.; Goetz, J.G.; et al. RAL-1 controls multivesicular body biogenesis and exosome secretion. J. Cell Biol. 2015, 211, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: a novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of RAB7A Protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Korkut, C.; Ataman, B.; Ramachandran, P.; Ashley, J.; Barria, R.; Gherbesi, N.; Budnik, V. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell 2009, 139, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.-A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles: messengers and mediators of tumor progression. Cell Cycle 2009, 8, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Bellingham, S.A.; Guo, B.B.; Coleman, B.M.; Hill, A.F. Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 2012, 3, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, N.; Dhawan, J. Exosomes: mobile platforms for targeted and synergistic signaling across cell boundaries. Cell Mol. Life Sci. 2017, 74, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2, 20677. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.G.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. El Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed]
- Newton, W.C.; Kim, J.W.; Luo, J.Z.Q.; Luo, L. Stem cell-derived exosomes: a novel vector for tissue repair and diabetic therapy. J. Mol. Endocrinol. 2017, 59, R155–R165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Guescini, M.; Guidolin, D.; Vallorani, L.; Casadei, L.; Gioacchini, A.M.; Tibollo, P.; Battistelli, M.; Falcieri, E.; Battistin, L.; Agnati, L.F.; et al. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp. Cell Res. 2010, 316, 1977–1984. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Helmig, S.; Frühbeis, C.; Krämer-Albers, E.-M.; Simon, P.; Tug, S. Release of bulk cell free DNA during physical exercise occurs independent of extracellular vesicles. Eur. J. Appl. Physiol. 2015, 115, 2271–2280. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: in sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Fracasso, F.; Pesce, V.; Cantatore, P.; Joseph, A.-M.; Leeuwenburgh, C.; Gadaleta, M.N.; Lezza, A.M.S. Age- and calorie restriction-related changes in rat brain mitochondrial DNA and TFAM binding. Age 2013, 35, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.-M.; Leeuwenburgh, C.; Lezza, A.M.S. Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS ONE 2013, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Pesce, V.; Fracasso, F.; Joseph, A.-M.; Leeuwenburgh, C.; Lezza, A.M.S. A comparison among the tissue-specific effects of aging and calorie restriction on TFAM amount and TFAM-binding activity to mtDNA in rat. Biochim. Biophys. Acta 2014, 1840, 2184–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. Why do we need autophagy? A cartoon depiction. Autophagy 2018, 14, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef] [PubMed]
- Martinvalet, D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudent, J.; McBride, H.M. The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr. Opin. Cell Biol. 2017, 47, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle Communication between Mitochondria and the Endolysosomal System. Front. Cell Dev. Biol. 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M.; et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions. Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [Green Version]
- Norambuena, A.; Wallrabe, H.; Cao, R.; Wang, D.B.; Silva, A.; Svindrych, Z.; Periasamy, A.; Hu, S.; Tanzi, R.E.; Kim, D.Y.; et al. A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-β oligomers. EMBO J. 2018, 37, e100241. [Google Scholar] [CrossRef]
- Norambuena, A.; Wallrabe, H.; McMahon, L.; Silva, A.; Swanson, E.; Khan, S.S.; Baerthlein, D.; Kodis, E.; Oddo, S.; Mandell, J.W.; et al. mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dement. 2017, 13, 152–167. [Google Scholar] [CrossRef]
- Assali, E.A.; Shlomo, D.; Zeng, J.; Taddeo, E.P.; Trudeau, K.M.; Erion, K.A.; Colby, A.H.; Grinstaff, M.W.; Liesa, M.; Las, G.; et al. Nanoparticle-mediated lysosomal reacidification restores mitochondrial turnover and function in β cells under lipotoxicity. FASEB J. 2018. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.F.; Tang, M.Y.; Fon, E.A.; Durcan, T.M. Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int. J. Biochem. Cell Biol. 2016, 79, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Desdín-Micó, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adh. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; Lorenzi, M.; Tanganelli, F.; Picca, A.; Bossola, M.; Menghi, A.; Bernabei, R.; Landi, F. Association between myocyte quality control signaling and sarcopenia in old hip-fractured patients: Results from the Sarcopenia in HIp FracTure (SHIFT) exploratory study. Exp Gerontol. 2016, 80, 1–5. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Eitan, E.; Green, J.; Bodogai, M.; Mode, N.A.; Bæk, R.; Jørgensen, M.M.; Freeman, D.W.; Witwer, K.W.; Zonderman, A.B.; Biragyn, A.; et al. Age-related changes in plasma extracellular vesicle characteristics and internalization by leukocytes. Sci. Rep. 2017, 7, 1342. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef] [PubMed]
- Alique, M.; Ruíz-Torres, M.P.; Bodega, G.; Noci, M.V.; Troyano, N.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Carracedo, J.; et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017, 9, 778–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weilner, S.; Keider, V.; Winter, M.; Harreither, E.; Salzer, B.; Weiss, F.; Schraml, E.; Messner, P.; Pietschmann, P.; Hildner, F.; et al. Vesicular Galectin-3 levels decrease with donor age and contribute to the reduced osteo-inductive potential of human plasma derived extracellular vesicles. Aging 2016, 8, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; Micolucci, L.; Cricca, M.; De Carolis, S.; Mensà, E.; Ceriello, A.; Procopio, A.D.; Bonafè, M.; Olivieri, F. Exosome-based immunomodulation during aging: A nano-perspective on inflamm-aging. Mech. Ageing Dev. 2017, 168, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; de Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Ding, W.; Guo, H.; Xu, C.; Wang, B.; Zhang, M.; Ding, F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 2016, 7, 17479–17491. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Zhang, W.; Jiao, B.; Pan, C.-Z.; Liu, X.; Shen, L. The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl. Neurodegener. 2017, 6, 3. [Google Scholar] [CrossRef]
- Tofaris, G.K. A Critical assessment of exosomes in the pathogenesis and stratification of Parkinson’s disease. J. Parkinsons Dis. 2017, 7, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front. Genet. 2012, 3, 297. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Sergeant, N.; Buée, L. Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer’s disease. Front. Physiol. 2012, 3, 229. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef]
- Jarmalavičiūtė, A.; Pivoriūnas, A. Exosomes as a potential novel therapeutic tools against neurodegenerative diseases. Pharmacol. Res. 2016, 113, 816–822. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.-C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018. [Google Scholar] [CrossRef] [PubMed]
- Anticoli, S.; Aricò, E.; Arenaccio, C.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Ferrantelli, F.; Lattanzi, L.; D’Urso, M.T.; Proietti, E.; et al. Engineered exosomes emerging from muscle cells break immune tolerance to HER2 in transgenic mice and induce antigen-specific CTLs upon challenge by human dendritic cells. J. Mol. Med. 2018, 96, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-C.; Tao, S.-C.; Dawn, H. Microfluidics-based on-a-chip systems for isolating and analysing extracellular vesicles. J. Extracell. Vesicles 2018, 7, 1508271. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 805. https://doi.org/10.3390/ijms20040805
Picca A, Guerra F, Calvani R, Bucci C, Lo Monaco MR, Bentivoglio AR, Coelho-Júnior HJ, Landi F, Bernabei R, Marzetti E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. International Journal of Molecular Sciences. 2019; 20(4):805. https://doi.org/10.3390/ijms20040805
Chicago/Turabian StylePicca, Anna, Flora Guerra, Riccardo Calvani, Cecilia Bucci, Maria Rita Lo Monaco, Anna Rita Bentivoglio, Hélio José Coelho-Júnior, Francesco Landi, Roberto Bernabei, and Emanuele Marzetti. 2019. "Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles" International Journal of Molecular Sciences 20, no. 4: 805. https://doi.org/10.3390/ijms20040805
APA StylePicca, A., Guerra, F., Calvani, R., Bucci, C., Lo Monaco, M. R., Bentivoglio, A. R., Coelho-Júnior, H. J., Landi, F., Bernabei, R., & Marzetti, E. (2019). Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. International Journal of Molecular Sciences, 20(4), 805. https://doi.org/10.3390/ijms20040805