1. Introduction
Fluid resuscitation with colloid and crystalloid solutions is one of the main interventions in the management of critically ill patients. Although the main role of volume replacement solutions lie in the restoration of intravascular volume, they have also proved to be potent modulators of inflammation at both the systemic and cellular levels, demonstrating either anti-inflammatory or pro-inflammatory effects [
1,
2]. Among the semisynthetic colloids, one of the most commonly used worldwide is hydroxyethyl starch (HES), which is produced by hydroxyethyl substitution of amylopectin. HES has shown an increased persistence in the intravascular space compared to other colloids [
3] and has been demonstrated to possess anti-inflammatory properties [
4,
5]. For instance, a wealth of evidence indicates that HES is able to modulate several leukocyte functions such as degranulation [
6], oxidative burst [
7,
8,
9,
10] and neutrophil–endothelium interaction [
11]. This last aspect is of particular relevance, considering the role of leukocytes in inducing microvascular permeability and endothelial damage [
12,
13]. Indeed, a large body of evidence suggests that neutrophil adhesion to the endothelium and their extravasation during inflammatory conditions might contribute to vascular leakage, either directly or through a paracrine pathway sustained by the release of granules [
12]. Thus, the control of the adhesion of neutrophils to endothelium might be beneficial in reducing organ dysfunction and vascular leakage [
4,
14]. On the other hand, a strong inhibition of adhesion in conditions where the role of neutrophils may be crucial, e.g. during infections, could worsen patient outcome [
15]. Previous studies showed that HES was able to impair leukocytes–endothelial interactions and neutrophil trans-endothelial migration. However, there are still conflicting results regarding the mechanisms by which HES can affect the leukocyte–endothelium couple, although a supposed inhibitory effect on neutrophil integrins had been advocated [
16,
17]. To address this question, in the present study we assessed the binding of HES to neutrophils and the effect of specific integrin-blocking antibodies on this interaction. Moreover, we evaluated the influence of HES on the chemotaxis of neutrophils in response to IL-8 and fMLP, in order to estimate the impact of HES-containing volume replacement solutions on neutrophils’ activity during in vitro simulated inflammatory conditions.
3. Discussion
The interaction between endothelial cells and neutrophils has a pivotal role in the early stages of acute inflammation. Thanks to this interaction, neutrophils can migrate towards the site of injury to eradicate noxious agents. However, when this process is massive and uncontrolled it can increase the already present vascular permeability, exacerbating vascular leak syndrome [
12,
23], a condition found in several critically ill patients. Thus, the control of neutrophil adhesion to the endothelium and of trans-endothelial migration may have large impact on clinical practice.
Previous studies showed that HES is able to impair the neutrophil–endothelial cell interaction. This occurs probably by inhibiting neutrophil integrin function, revealed by a decrease in firm adhesion and trans-endothelial migration without affecting rolling efficiency [
16,
17]. On the other hand, other studies suggested that HES decreased neutrophil adhesion through an endothelial-dependent mechanism, since only the treatment of endothelial cells with HES was found to affect adhesion of leukocytes, leuko-aggregation and rolling velocity [
11,
24], along with decreased endothelial activation [
25]. Thus, currently published data on the topic are still highly conflicting and, overall, lack direct evidence of the binding of HES to neutrophils, which is supportive of any of the aforementioned mechanistic hypotheses.
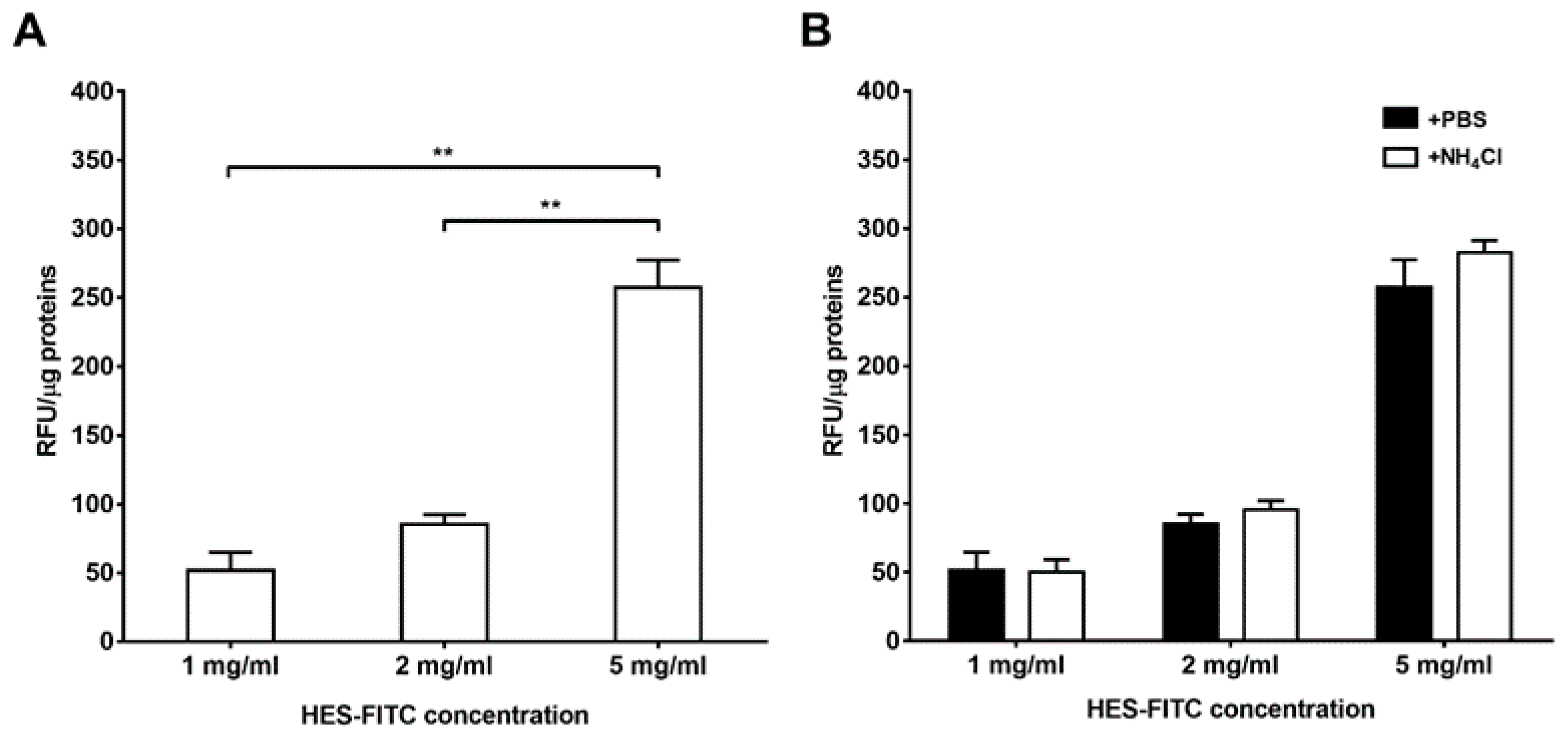
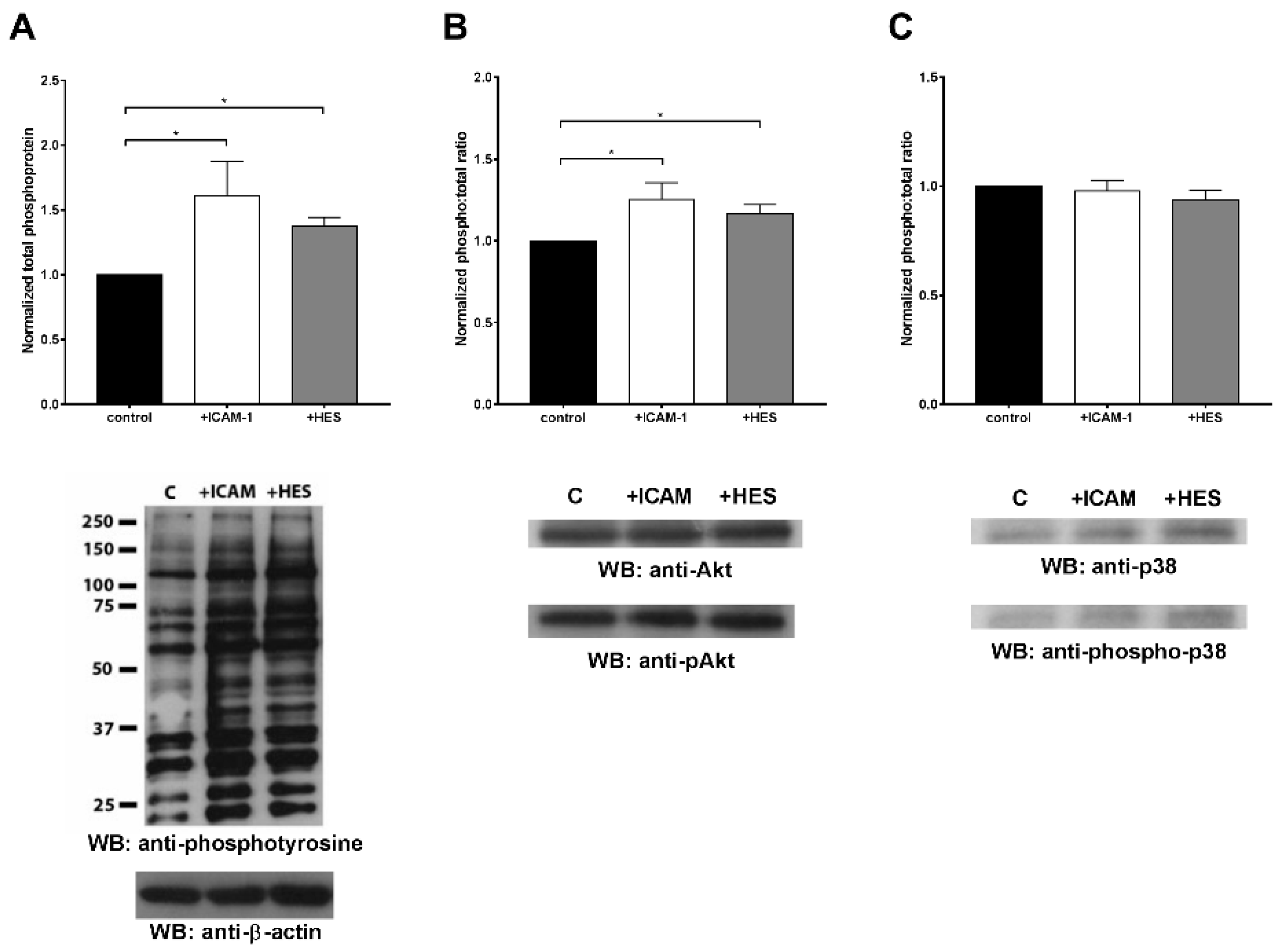
Owing these premises, our study aimed to evaluate in vitro the physical interaction between HES and neutrophils, and to investigate the influence of HES on neutrophil chemotaxis towards different stimuli as well as the players involved in HES binding. As highlighted by our results, for the first time we were able to directly demonstrate that HES is able to solely bind to the extracellular side of the plasma membrane without being internalized. Indeed, by using a fluorescently labelled HES (HES-FITC) we observed increasing cell-associated fluorescence with increasing HES-FITC concentration, fluorescence that was completely abolished upon treatment with a quenching agent. In addition, treatment with ammonium chloride, a lysosomotropic agent, did not exert any effect on FITC fluorescence, therefore confirming that HES is not internalized into phagosomes.
It is commonly accepted that integrins exist on circulating cells in a dynamic equilibrium between three states with different ligand affinities: low, intermediate, and high [
22]. Several studies reported that the ligation of integrins with substrates shifts this equilibrium towards the high-affinity state starting the outside-in signaling cascade, leading to phosphorylation of intracellular proteins and various effects. Therefore, from our data we can hypothesize that HES may behave as a physiological substrate for integrins by shifting the equilibrium towards the high-affinity state, further demonstrating its ability to bind to resting cells. Moreover, this binding seems sufficient to trigger outside-in signaling, as evidenced by the increased total phosphorylation of tyrosine found in intracellular proteins, events that can be further translated into the activation of signaling cascades modulating neutrophil responses. Indeed, from our results we observed that HES is able to activate the PI3K/Akt pathway, leaving p38/MAPK unaffected. The activation of Akt was previously associated with the modulation of lifespan [
22] and oxidative burst [
26] of neutrophils. However, the specific effect of HES on the activation of these cells is still a matter of debate and inconclusive. Therefore, our results suggested that the beneficial effect of HES observed in other studies may be at least partially dependent on neutrophil-associated mechanisms [
16,
17]. This is also in line with recent observations from Rossaint et al. on an animal model where HES was able to decrease inflammation, neutrophil recruitment in several organs, and their activation as measured by extracellular trap formation [
27]. Nonetheless, we cannot completely exclude that the anti-inflammatory effects of HES [
27,
28] may be mediated by mechanisms also involving endothelial cells. In fact, it has been shown that HES can be internalized through pinocytosis by HUVEC (Human Umbilical Vein Endothelial Cell) [
29], although no firm binding of the starch on the surface of these cells has been observed. Furthermore, a decreased expression of adhesion molecules, in particular E-selectin, on endothelial cells after treatment with HES has been reported [
11,
30]. However, contrasting findings have been also reported [
16,
17,
29].
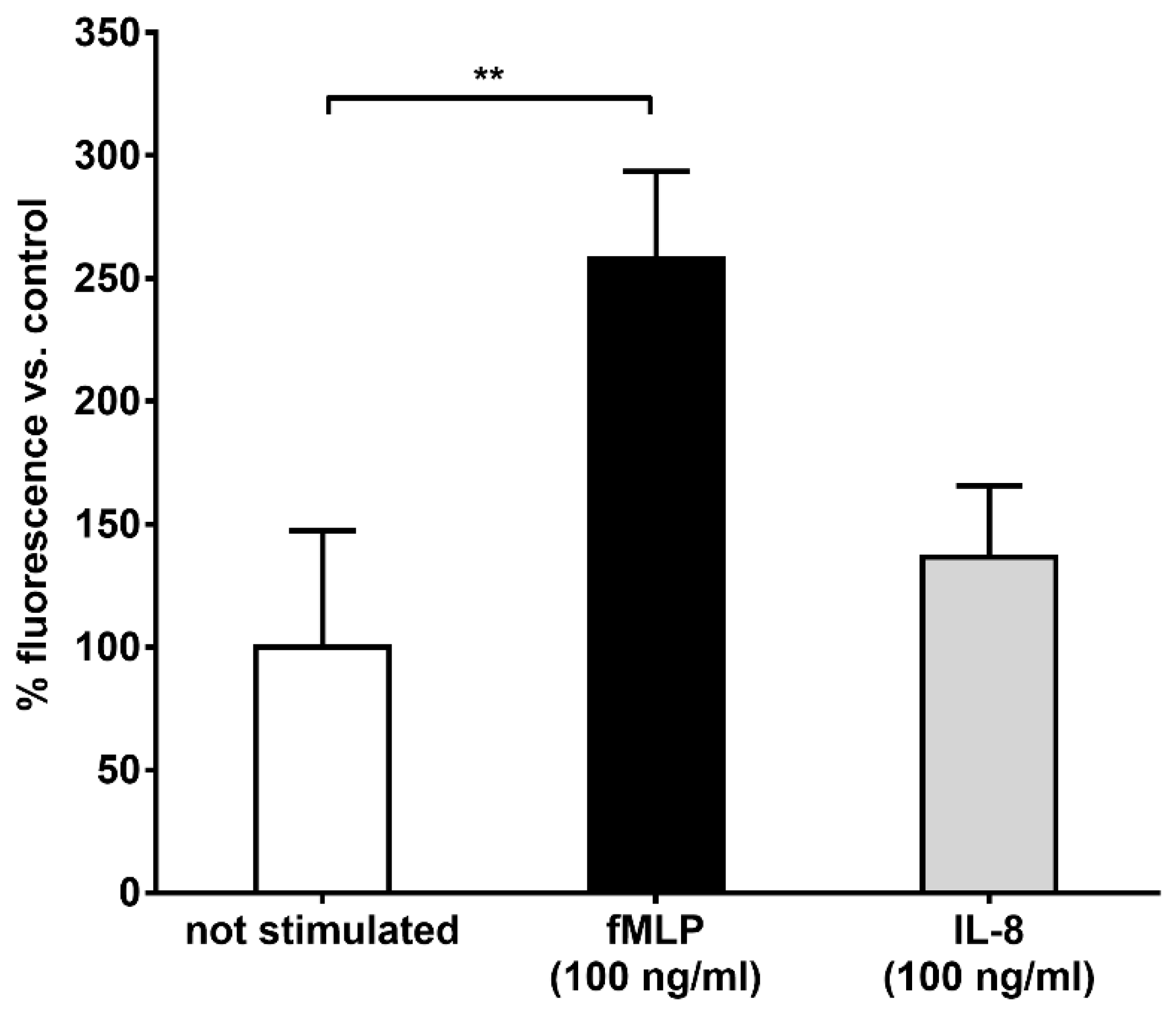
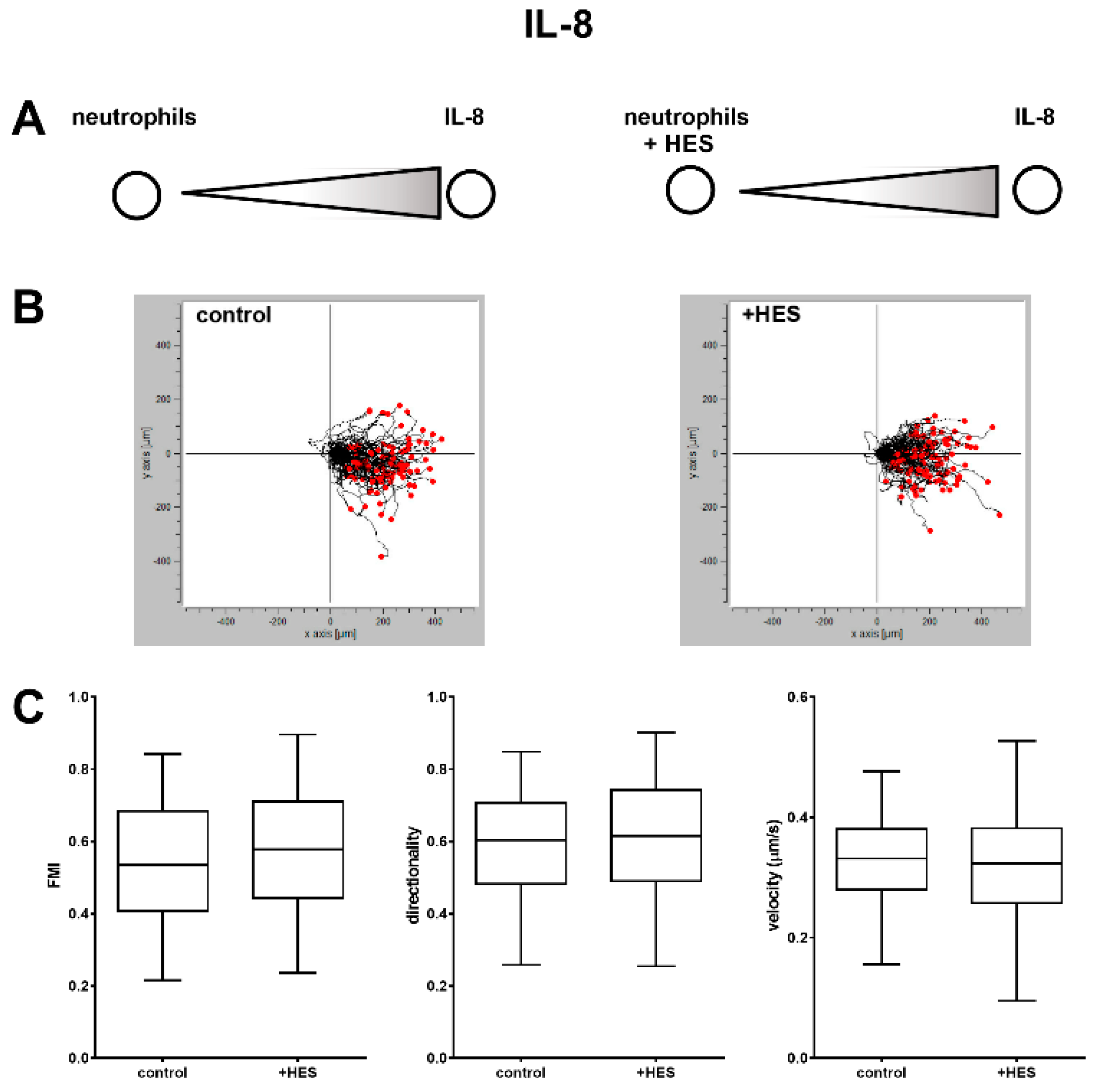
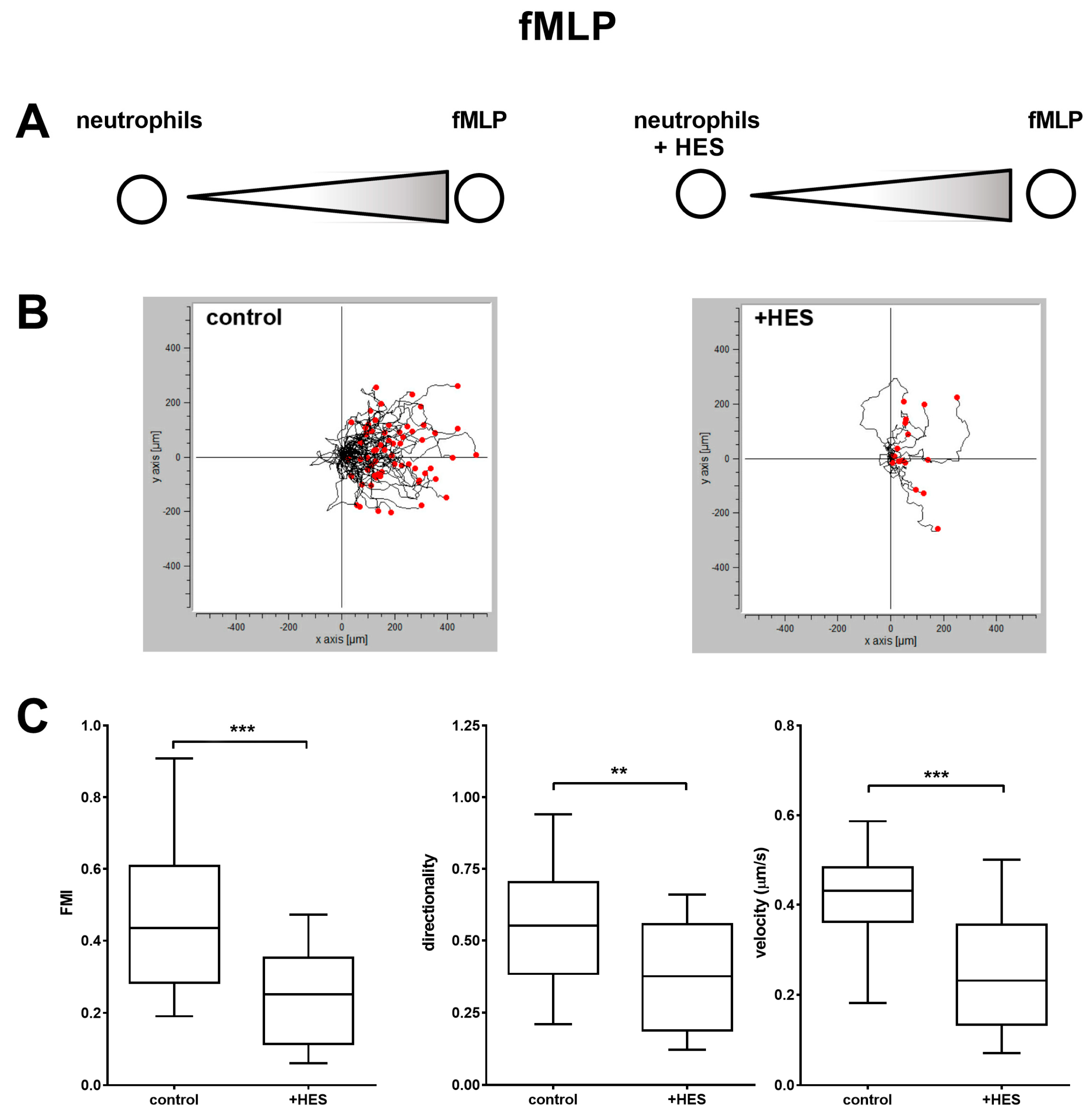
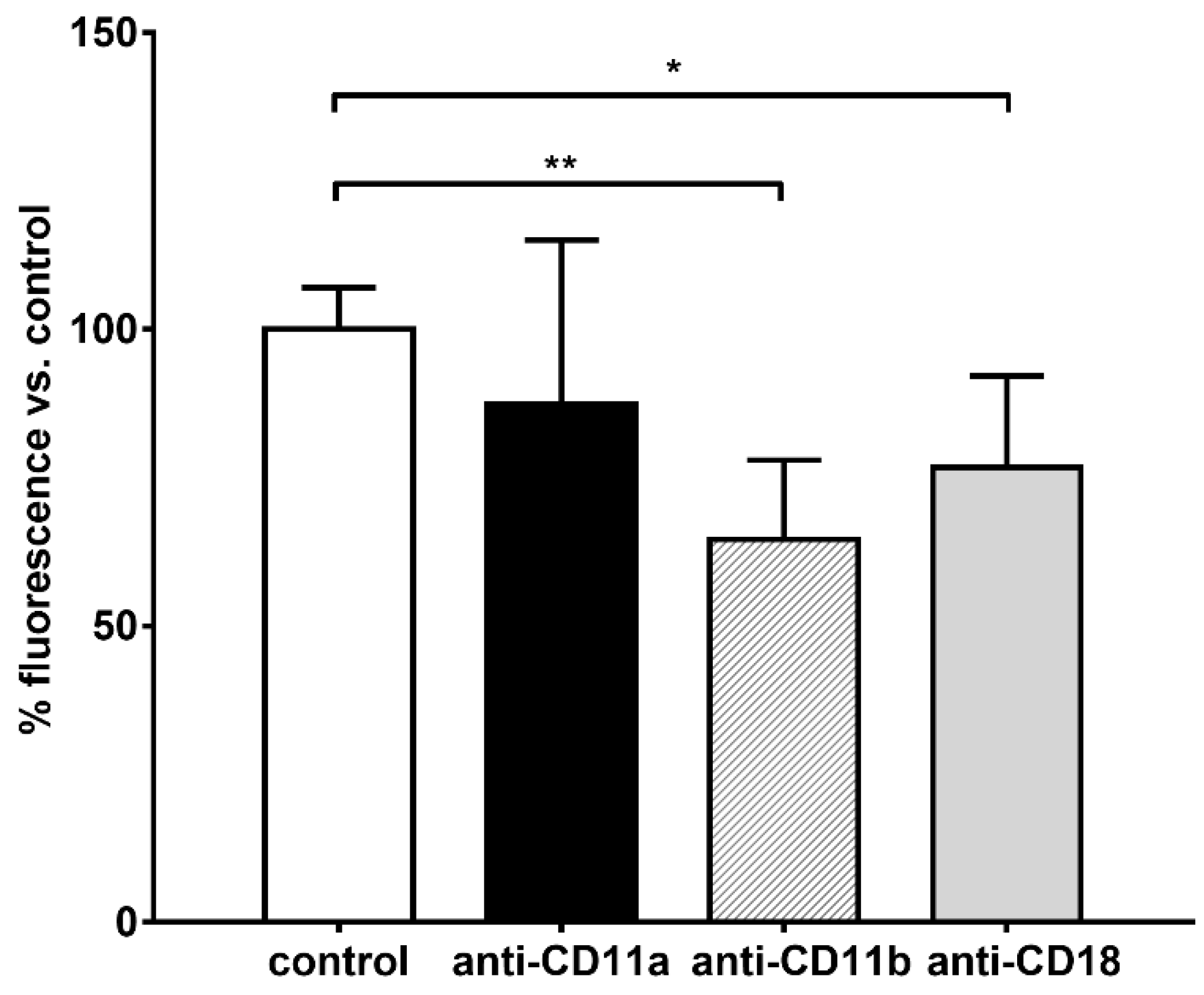
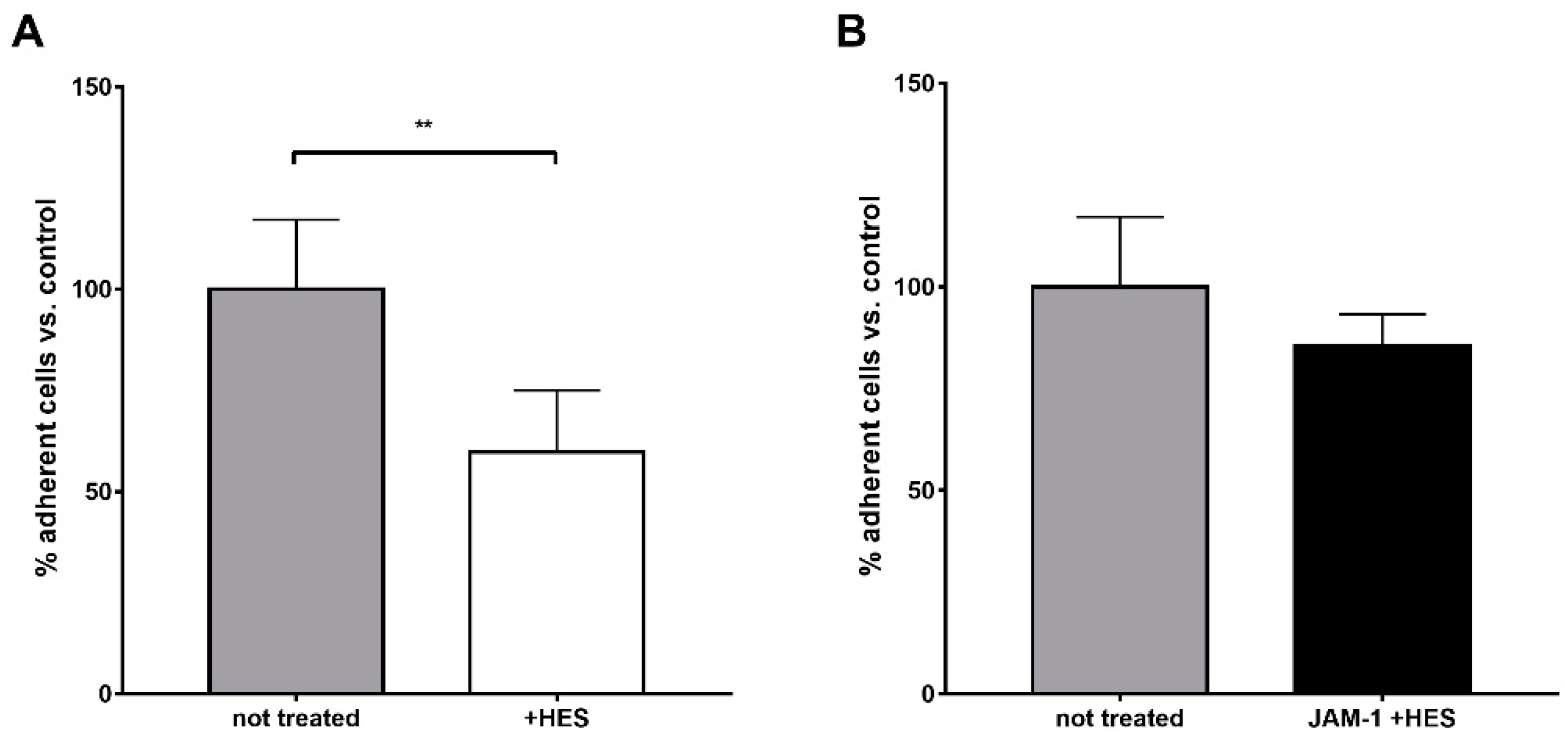
Differently, the body of literature dealing with the interaction between HES and neutrophils is highly consistent and points to the significant impact of synthetic colloids on the functionality of these immune cells. Besides the previous interference on degranulation and bacteria killing capacity [
6,
8], our results confirmed that chemotaxis is also massively impaired, with a striking decrease in cell migration (FMI, velocity and directionality) only towards “end-target” chemoattractants (e.g. fMLP) but not IL-8. As revealed by our results, the decrease in both FMI and velocity are mainly due to a disruption of Mac-1 (CD11b/CD18) action. Indeed, we observed that blocking the CD11b and, to a lesser extent, the CD18 subunits with specific antibodies was able to prevent the binding of HES-FITC to the cell surface with a supposed cooperative mechanism, since the cumulative percentage decrease in fluorescence intensity was almost 80%, whereas blocking the CD11a subunit did not exert any effect. This result is also supported by the finding of a significant increase in fluorescence intensity after treatment with fMLP but not IL-8, stimuli that can differently affect the affinity of the two integrins for their ligands with an increased avidity of Mac-1 following fMLP stimulation [
31]. Moreover, this was further confirmed by the finding of an impaired adhesion on fibrinogen, a CD11b specific substrate [
20], without affecting adhesion to JAM-1, a preferential substrate of CD11a [
21]. Of note, the impairment in directionality we observed in cell migration towards fMLP after treatment with HES could be due to the interaction of HES with alpha 4-integrins as well. Indeed, it is known that directionality in neutrophil chemotaxis is mediated by the action of alpha-4 and LFA-1 integrins [
32]; since we did not observe significant binding of HES to LFA-1, the only possible interactor remains alpha-4 integrin. However, without further experiments this remains mere speculation.
Collectively, our results might help to explain previous data showing an alteration in the rate of trans-endothelial migration of neutrophils treated with HES without impairing their rolling on activated endothelial cells. Indeed, while rolling is mainly mediated by selectins [
33], the two integrins expressed on neutrophils’ surfaces serve different functions during the migration cascade: on one side, LFA-1 seems to be more involved in early adhesion strengthening to the endothelium [
34]; on the other side, Mac-1 is essential for the intraluminal crawling of neutrophils to emigration sites [
35]. Thus, by preventing the binding of Mac-1 to its ligand, HES may be able to alter intraluminal crawling, controlling neutrophil migration to sites of inflammation. However, the observed lack of binding of HES to LFA-1 disagreed with the decreased tethering rate from other studies [
16,
17], although this might be due to differences in the employed techniques. In fact, only flow-chamber assays can be used to study neutrophil arrest and trans-endothelial migration in vitro, whereas the assays we used do not mimic dynamic conditions such as blood flow.
Nonetheless, the specific effect we observed may have a large impact on clinical practice, especially when a low activation of neutrophils may be advantageous, such as during sterile inflammation [
33] and ischemia/reperfusion injury [
36,
37], where the blocking of integrin activation has proven to be effective in preventing tissue damage [
38]. On the other hand, when perfect functionality of neutrophils are mandatory, such as in the context of bacterial inflammation, the perturbation of the adhesion cascade may be more harmful than beneficial [
27].
This study was not without limitations. First, the lack of data on the affinity of Mac-1 and LFA-1 for HES in an isolated system and different activation states of integrins. Second, the use of the “static” under agarose assay instead of a flow-chamber assay may have weakened our data, although this is the first study identifying Mac-1 as the interacting partner of HES. Third, we did not perform experiments on activated endothelial cells to confirm/deny that HES firmly binds to these cells.
In conclusion, the binding of HES to the extracellular side of the plasma membrane of neutrophils and the consequent impairment of chemotaxis can be pictured as an anti-inflammatory property of the molecule. This is of particular interest, considering the long persistence of HES in the extravascular space. Thus, the observed effect can be largely dependent on its vascular elimination and may have an impact on the choice of volume replacement solution in clinical settings. Therefore, the type of inflammatory condition of patients should be an additional parameter to consider in the choice of the volume replacement strategy in order to exploit the possible immunomodulatory properties of hydroxyethyl starch. Further studies are necessary to determine the real clinical relevance of our findings.
4. Materials and Methods
4.1. Reagents
Ficoll-Hypaque Plus was purchased from GE Healthcare (Milan, Italy; Cat. No. 17-1440-02). Dextran 500 (Cat. No. 31392), FITC isomer I (Cat. No. F7250), ethanol absolute (Cat. No. 02860), fMLP (Cat. No. F3506), agarose (Cat. No. A9045), RPMI-1640 (Cat. No. R6504), bovine serum albumin (BSA, Cat. No. A8022), bicinchoninic acid assay kit (Cat. No. BCA1) were purchased from Sigma-Aldrich (Milan, Italy). HES (Tetraspan) was obtained from BBraun (Melsungen, Germany). Fetal bovine serum (FBS) was purchased from Immunological Sciences (Rome, Italy; Cat. No. EU-000-500). DMSO (dimethyl sulfoxide) was purchased from Merck (Darmstadt, Germany; Cat. No. 1.09678.0100). IL-8 was purchased from Peptrotech (London, UK; Cat. No. 200-08M). Anti-CD11a (LFA-1; Cat. No. an3981), anti-CD11b (MAC-1; Cat. No. ab130428), and anti-CD18 (Cat. No. ab8220) blocking antibodies were purchased from Abcam (Cambridge, UK) and isotypic antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). Anti pAkt1/2/3 (Cat. No. sc515451), total Akt1/2/3 (Cat. No. sc81434), phosphorylated p38 (Cat. No. sc166182) and total p38 (Cat. No. sc33688) were from Santa Cruz Biotechnology (Dallas, TX, USA).
4.2. Labeling of HES with Fluorescein Isothiocyanate (FITC)
The labeling of HES with FITC was carried out according to the method proposed by Ständer and coworkers [
39], with some modifications. Briefly, 0.1 g of HES was dissolved in 1.2 mL of DMSO and heated at 95 °C. After the complete dissolution of HES, 10 mg of FITC were added to the mixture and incubated for 6 h at 95 °C. The solution was cooled to room temperature and was added to 8 mL of absolute ethanol to precipitate the labeled HES. After a centrifugation of 10 min at 3300 rpm the supernatant was discarded and the pellet suspended in 1.6 mL of distilled water and dialyzed extensively against water. The final isolation of the labeled HES was done by freeze drying.
4.3. Isolation of Neutrophils and Evaluation of the Binding of HES to Their Plasma Membrane
Buffy coats were collected from the Blood Bank of S. Anna Hospital, Ferrara. All the data were analyzed anonymously, and the authors did not have any sensitive information about the participants. Neutrophils were isolated by gradient centrifugation and dextran sedimentation as previously described [
6]. The cells were further suspended in HBSS (5.3 mM KCl, 0.44 mM KH
2PO
4, 138 mM NaCl, 0.3 mM Na
2HPO
4, 10 mM HEPES, 5.6 mM Glucose, pH 7.4) without calcium and magnesium and kept on ice until use.
In order to evaluate the binding of HES to the plasma membrane of neutrophils, the cells were first treated with different concentrations of HES-FITC and then, after washing steps, the cell-associated fluorescence intensity was measured. Cells were further treated with ammonium chloride (NH
4Cl), a lysosomotropic agent that penetrate in the cells raising the pH of the phagolysosome [
40] thus enhancing the fluorescence of the FITC. Finally, to confirm the association of HES to the outer plasma membrane, the cells were treated with trypan blue in order to quench the extracellular fluorochrome emission [
40].
Briefly, freshly-isolated neutrophils (8 × 10
6 cells) were suspended in HBSS containing different concentrations of HES-FITC (1 mg/mL, 2 mg/mL or 5 mg/mL) dissolved in HBSS with 1 mM of CaCl
2 and MgCl
2, (herein referred to as complete HBSS) and incubated for 15 min at 37 °C. The highest concentration of HES (5 mg/mL) resembles the plasma concentration observed within the first 6 h after a bolus infusion of 500–1000 mL of the starch [
16]. After incubation, cells were centrifuged for 10 min at 1100 rpm at 4 °C and washed twice with PBS. Finally, the pellets were suspended in 1 mL of complete HBSS and 100 µL of suspension (800,000 cells) were dispensed in quadruplicate in a clear flat bottomed 96 wells microtiter plate (Greiner Bio-One, Cat. No. 655101) and the fluorescence of FITC was read with a microplate fluorimeter (Tecan Infinite M200, Austria) at λ
ecc = 490 nm and λ
em = 520 nm. After the reading, 50 µL of PBS were added into two wells and 50 µL of 150 mM ammonium chloride (NH
4Cl, 50 mM final concentration) were added to the remaining wells and the fluorescence was measured again.
To quench the signal coming from the plasma membrane, neutrophils (8 × 106 cells) were suspended in 1 ml of acetate buffer (20 mM acetate buffer, pH 5.8 containing 130 mM NaCl) and 100 µL of suspension (800,000 cells) were dispensed in quadruplicate in a clear flat bottomed 96 wells microtiter plate and the fluorescence of FITC read with a microplate fluorimeter. The remaining cells were incubated with Trypan Blue dissolved in acetate buffer at a final concentration of 0.2 mg/mL for 20 s at room temperature. Cells were then washed twice with acetate buffer and suspended in the same buffer. Finally, 100 µL of suspension (800,000 cells) were dispensed in quadruplicate and the resulting fluorescence read with a microplate fluorimeter.
4.4. Binding of HES to the Plasma Membrane of Stimulated Neutrophils
Neutrophils (8 × 106 cells) were suspended in complete HBSS containing 5 mg/mL HES-FITC and 100 ng/mL fMLP, 100 ng/mL IL-8 or without stimulation and incubated for 15 min at 37 °C. At the end of the incubation, the neutrophils were centrifuged at 1100 rpm at 4 °C for 10 min and washed twice with PBS. The cells were then suspended in 1 mL of complete HBSS and 100 µL of suspension (800,000 cells) were dispensed in quadruplicate in a clear flat bottomed 96 wells microtiter plate, and the fluorescence of FITC was read with a microplate fluorimeter.
4.5. Binding of HES after Treatment of Neutrophils with Integrin-Blocking Antibodies
Neutrophils (1 × 106 cells) were suspended in complete HBSS and incubated for 30 min at room temperature with 20 µg/mL (corresponding to 2 µg of antibodies per million of cells) of anti-CD11a (LFA-1, Abcam, Cat. No. AB3981, clone MEM-83), anti-CD11b (Mac-1, Abcam, Cat. No. AB130428, clone ICRF44), anti-CD18 (Abcam, Cat. No. AB8220, clone MEM-148) blocking antibodies, or with isotypic antibodies as a control. Afterwards, the cells were centrifuged at 1100 rpm for 5 min, treated with 5 mg/mL HES-FITC dissolved in complete HBSS. Cells were then incubated for 15 min at room temperature in the dark. Finally, the cells were washed twice with PBS and suspended with 125 µL of PBS and 100 µL of suspension (~800,000 cells) were dispensed in a clear flat bottomed 96-well microtiter plate and the fluorescence of FITC read with a microplate fluorimeter.
4.6. Measurement of Total Proteins with Bicinchoninic Acid
In order to account for possible differences in the number of cells loaded in the wells, we determined the total proteins content per well and the values of fluorescence intensity throughout the paper were expressed as RFU/µg of total protein. Briefly, the cells were lysed with 1% Triton X-100 (final concentration) for 30 min at 4 °C and 25 µL of lysate or standard (Bovine Serum Albumin, BSA, in the range 0.25–1 mg/mL) were dispensed in a flat bottomed microtiter plate and the total proteins content was measured with the Bicinchoninic Acid Assay by using a commercially available kit (Sigma-Aldrich, Milan, Italy, Cat. No. BCA1), according to the manufacturer instructions.
4.7. Under-Agarose Assay
Neutrophil chemotaxis was evaluated by using the under agarose assay essentially as described elsewhere [
41]. Briefly, Petri dishes (35 × 10 mm) were coated with 10% FBS in PBS for 30 min at room temperature and washed twice with PBS. Then, the plates were filled with 3 mL of a 0.45% agarose dissolved in 50% HEPES-buffered complete HBSS and 50% supplemented with 20% FBS. The agarose was then allowed to solidify and two wells 3.5 mm diameter and 2.2 mm apart were cut into each gel. The gels were then equilibrated at 37 °C for 1 h and 10 µL of chemokine (1 pmol of fMLP or 10 pmol of IL-8) were loaded in the outer well, whereas 10 µL of 1 × 10
7 cells/mL (1 × 10
5 cells) not treated or pretreated for 15 min with 5 mg/mL of HES dissolved in complete HBSS were loaded in the inner well. After 1.5 h of incubation at 37 °C, images of migrating neutrophils were acquired every 20 s for 20 min, with a Canon EOS 700D digital reflex attached to an inverted microscope (Olympus IMT-2) equipped with a thermostatic chamber. The tracks of the migrating cells were then acquired by using ImageJ with multitracker plugin and the coordinates of the cells were analyzed by using a free software (Ibidi Chemotaxis and Migration Tool), in order to obtain quantitative parameters of migration such as FMI (forward migration index, parallel to the gradient and calculated as the ratio between the x coordinate of the cell’s end point of migration and the total distance accumulated), directionality (representing a measurement of the directness of cell trajectories, the ratio of the Euclidian distance and the total accumulated distance of a cell) and velocity (displacement/time).
4.8. Static Adhesion Assay on Fibrinogen and JAM-1
To evaluate the in vitro adhesion of neutrophils on substrates such as fibrinogen and JAM-1 (Junction Adhesion Molecule-1) in the absence or presence of HES, the cells were washed twice with Ca-Mg-free HBSS and then fluorescently labeled by incubation with 3 µM of calcein-AM (Sigma-Aldrich, cat. No. C1359) diluted in Ca-Mg-free HBSS for 30 min at 37 °C. The cells were centrifuged 5 min at 400× g, washed twice with Ca-Mg-free HBSS, and then suspended in complete HBSS and treated/not treated with 5 mg/mL HES for 15 min at 37 °C. The cells were further centrifuged 5 min at 400× g, washed once with complete HBSS and suspended in the same buffer at the concentration of 1 × 106 cells/mL. Finally, 50 µL of cell suspension (corresponding to 50,000 cells) were dispensed in wells of a 96-well plate ELISA plate (Nunc MaxiSorp, Thermofisher Scientific, Monza, Italy; Cat. No. 44-2404-21) pre-coated with fibrinogen (Sigma-Aldrich, Milan, Italy; cat. No. F3879) or JAM-1 (AbCam, Cambridge, UK; cat. No. ab132180). Coating was performed overnight at 4 °C by dispensing 50 µL of protein prepared at the final concentration of 10 µg/mL in modified-PBS (pH 7.4, containing 1 mM CaCl2 and 2 mM MgCl2). After one washing step with modified-PBS, the wells were incubated with 100 µL of 1% BSA dissolved in modified-PBS for 1 h at room temperature. At the end of the incubation, the wells were washed one time with modified-PBS and 50 µL of labeled cell suspension were dispensed in the wells. After 30 min of incubation at 37 °C, the total fluorescence was read (excitation: 490 nm; emission 525 nm), giving the pre-wash fluorescence. The wells were then gently washed three times with modified-PBS and fluorescence per well was measured again, giving the post-wash fluorescence. The ratio between the post-wash and pre-wash fluorescence multiplied by 100 represented the percentage of adherent cells per well.
4.9. Western Blot Analysis of Total Phospo-Tyrosine, Akt1/2/3 and p38
The activation of integrins by HES was evaluated by Western blot analysis of total phospho-Tyrosine on cells treated with HES, ICAM-1 or not treated as a control. In addition, we evaluated specific pathways activation by determining Akt1/2/3 and p38 phosphorylation status. Briefly, 2 × 106 cells were incubated in complete HBSS without stimuli, in the presence of 5 mg/ml of HES or 10 µg/mL of ICAM-1 (Sigma-Aldrich, cat. No. SRP3057) for 15 min at 37 °C. Cells were then centrifuged for 5 min at 400× g at 4 °C, washed once in PBS and then incubated for 30 min at 4 °C in lysis buffer containing phosphatase and protease inhibitors (50 mM Tris, pH 7.6, 150 mM NaCl, 25 mM β-Glycerophosphate, 50 mM NaF, 2 mM Na3VO4, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100, Halt protease Inhibitor cocktail, Thermo Scientific, Cat. No. 87786). Total protein content was evaluated by the Bradford assay and 20 µg of proteins were loaded onto a 10% sodium dodecyl sulphate polyacrylamide gel (SDS-PAGE) under reducing conditions and electrophoresed for 45 min at 35 mA constant amperage. The separated proteins were then transferred at 300 mA for 90 min to a 0.45 µm-pore PVDF (Polyvinylidene fluoride) membrane (Immobilon-P, Merck, Darmstadt, Germany; Cat. No. IPVH00010) and the non-specific sites were blocked by incubating the membranes for 1 h at room temperature with 5% BSA (Sigma-Aldrich, Fraction V, Cat. No. 05482) in TBS-T (Tris Buffered Saline, pH 7.4, 0.1% Tween-20). After washing steps in TBS-T, the membrane was incubated overnight at 4 °C with either anti-phospho-Tyrosine (Elabscience, Huston, Texas, USA; Cat. No. E-AB-21335, diluted 1:2000), anti-beta-actin (Sigma-Aldrich, Milan, Italy; Cat. No. A3854, diluted 1:100,000), pAkt1/2/3 (1:2000), total Akt1/2/3 (1:2000), p-p38 (1:2000) or total p38 (1:2000) antibodies. Afterwards, the membrane was washed three times with TBS-T, incubated with the proper secondary antibody (diluted 1:100,000 in blocking buffer) for one hour at room temperature and washed three times each with TBS-T and TBS. Finally, the bands were revealed by ECL system (SuperSignal West Femto, Thermo Fisher Scientific, Monza, Italy; Cat. No. 34095) and film exposure. Band intensity analysis was carried out by using the program ImageStudio Lite v5.2 (Li-Cor Biosciences, Lincoln, NE, USA) and expressed as ratio relative to the loading control (beta-actin or the total form of Akt1/2/3 or p38) and normalized by the not treated condition.
4.10. Statistical Analysis
The comparisons of fluorescence between groups and the percentage of adherent cells on different substrates were performed by t-test or by 1-way ANOVA followed by Sidak’s multiple comparison test. The comparisons of the migration parameters between the HES-treated cells and the control were performed by Mann-Whitney U test. The comparisons between the normalized band intensities from western blots were performed by t-tests. All the analyses were carried out with GraphPad® Prism 6 and a p < 0.05 was considered significant.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}