Reactivity of Gold(I) Monocarbene Complexes with Protein Targets: A Theoretical Study




Abstract
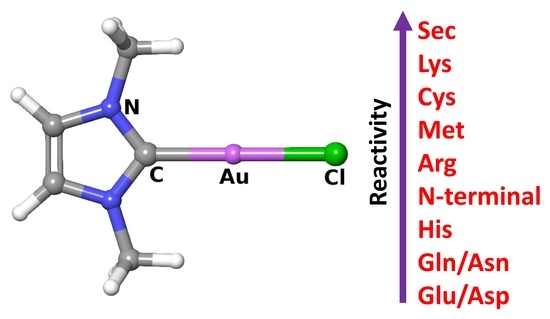
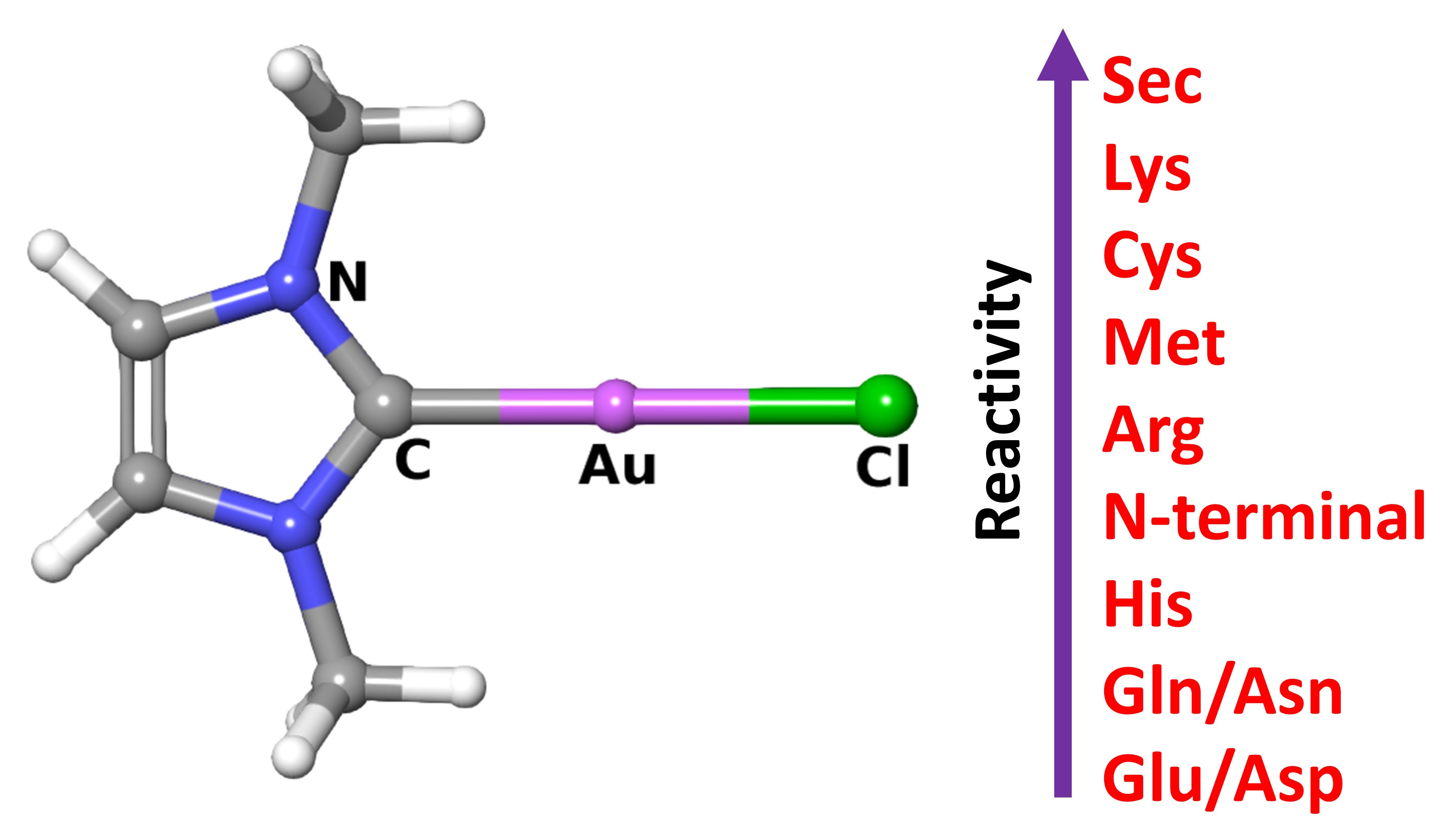
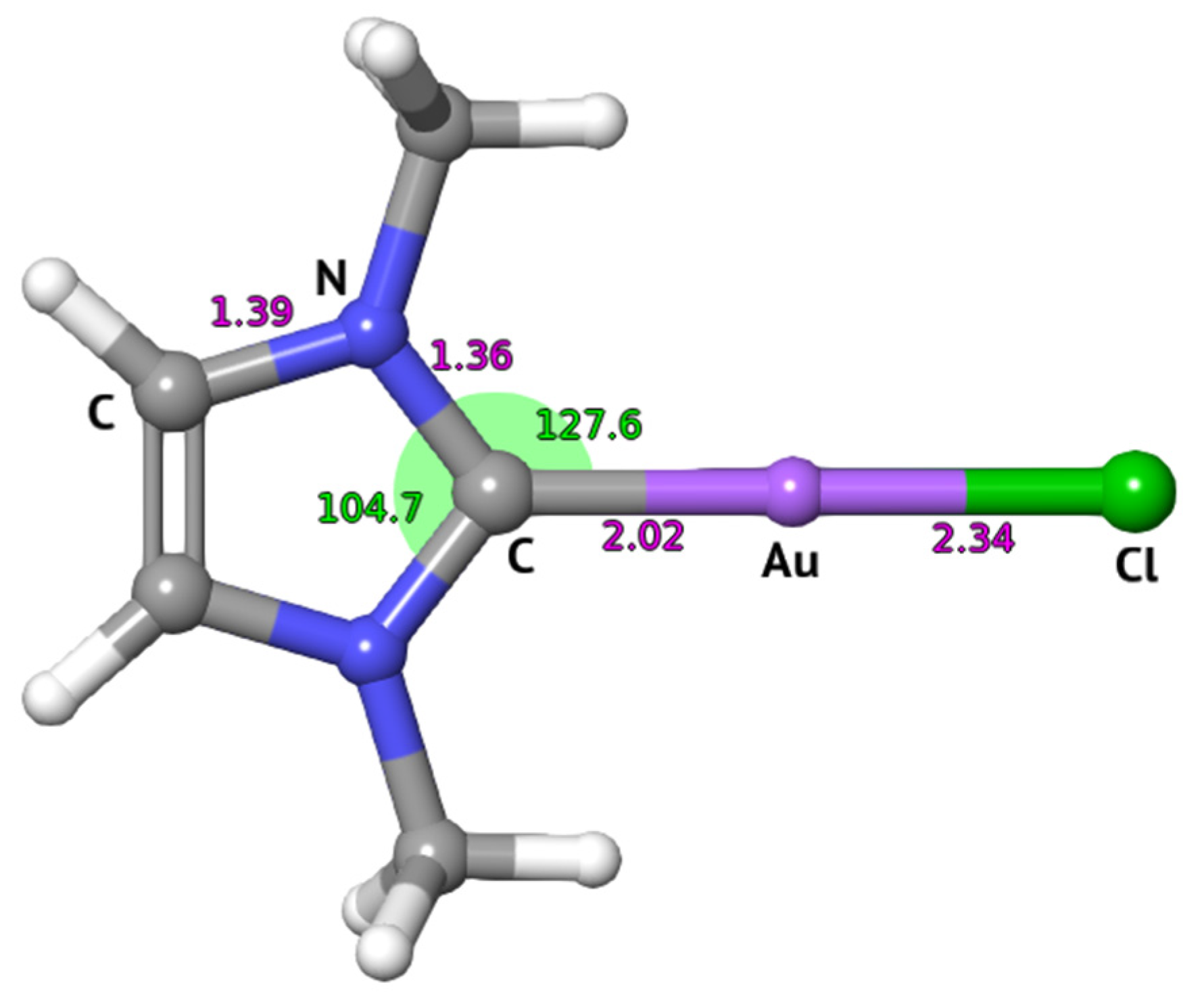
:
1. Introduction
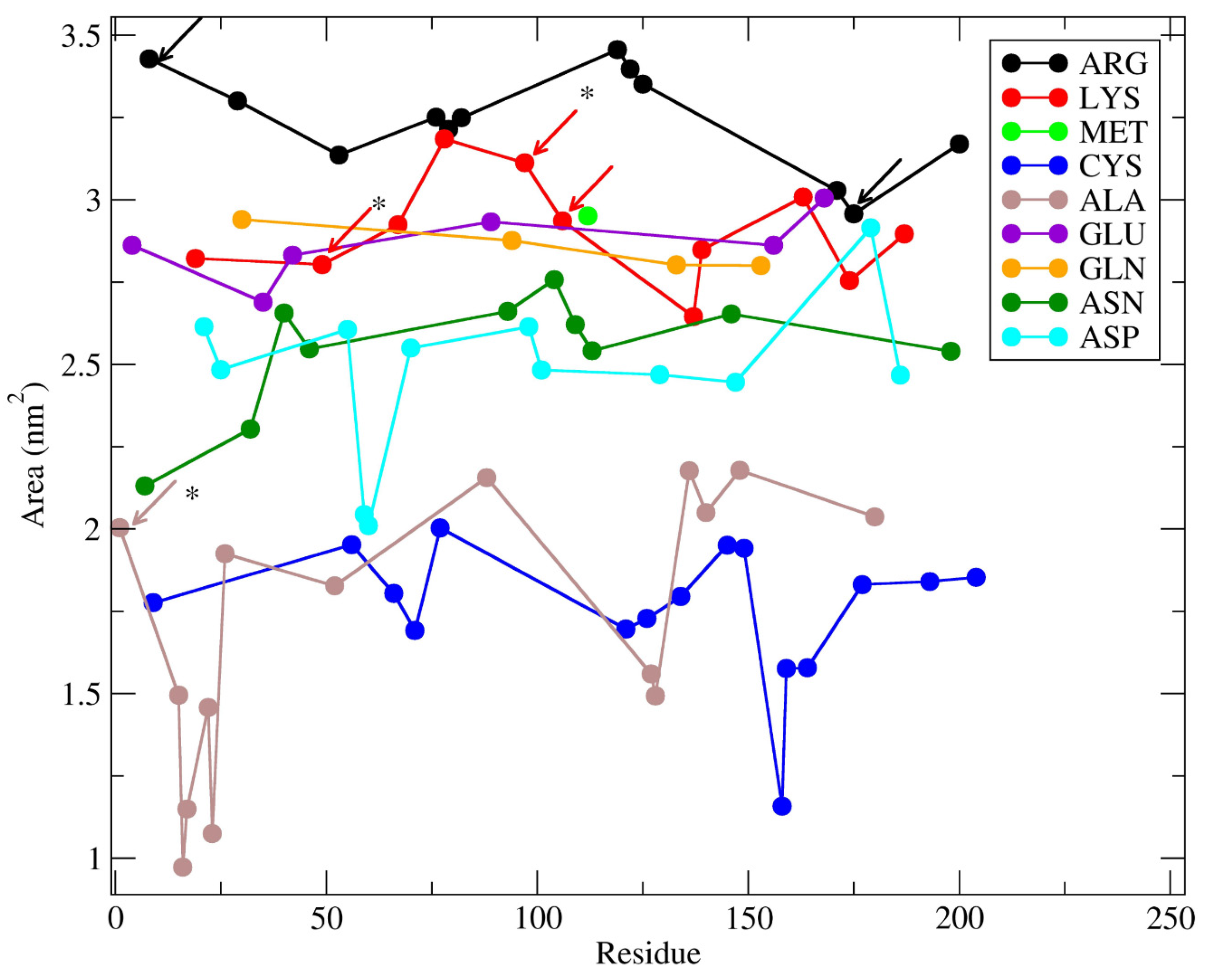
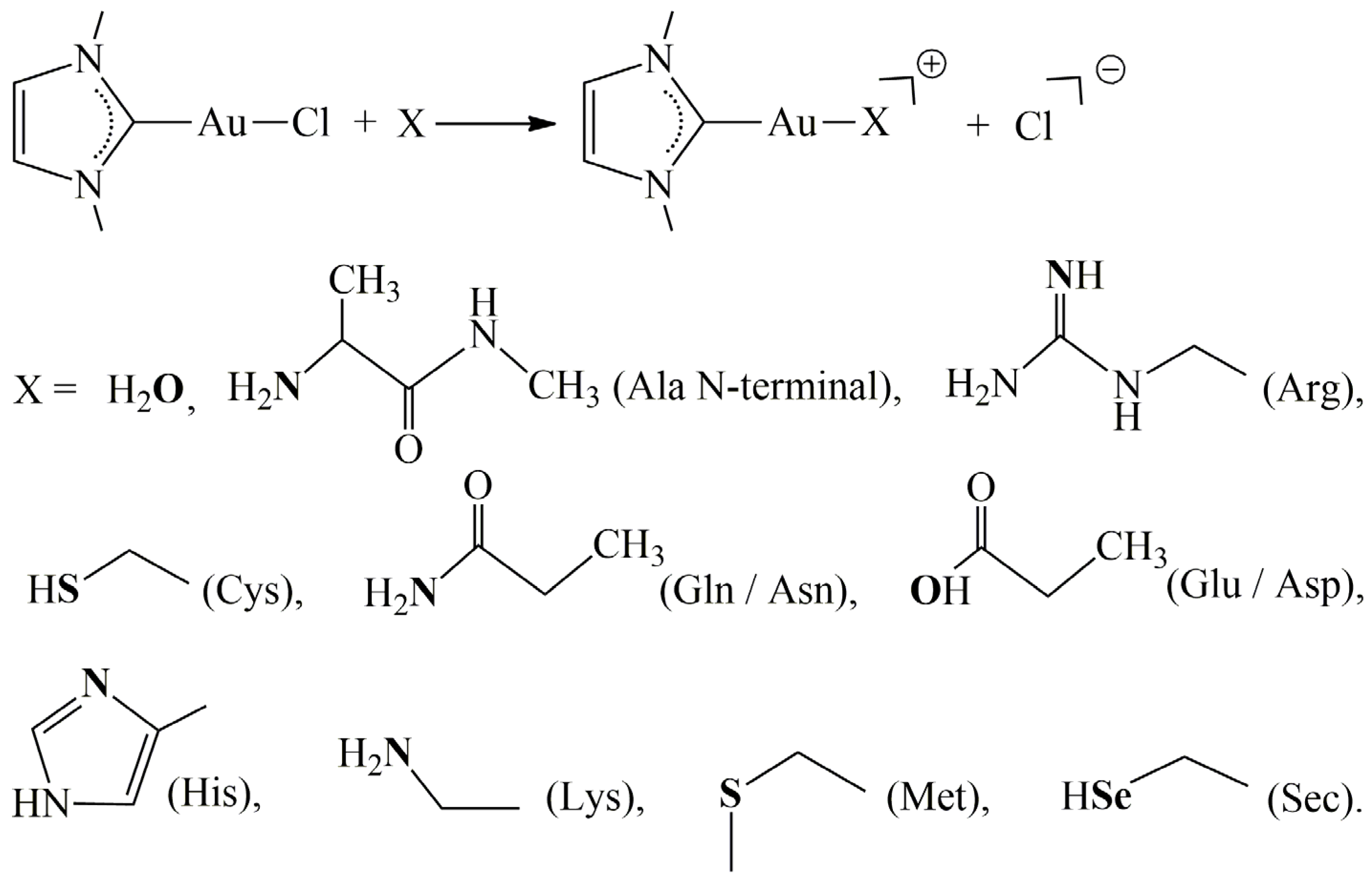
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| PB | Poisson–Boltzmann |
| MP2 | Møller–Plesset 2nd order perturbation theory |
| NHC | N-Heterocyclic Carbenes |
References
- Bindoli, A.; Rigobello, M.P.; Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Thioredoxin reductase: A target for gold compounds acting as potential anticancer drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar] [CrossRef]
- Milacic, V.; Dou, Q.P. The tumor proteasome as a novel target for gold (III) complexes: Implications for breast cancer therapy. Coord. Chem. Rev. 2009, 253, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Molter, A.; Mohr, F. Gold complexes containing organoselenium and organotellurium ligands. Coord. Chem. Rev. 2010, 254, 19–45. [Google Scholar] [CrossRef]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Filipovska, A. Gold compounds as therapeutic agents for human diseases. Metallomics 2011, 3, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef] [PubMed]
- Che, C.M.; Sun, R.W.Y. Therapeutic applications of gold complexes: Lipophilic gold (III) cations and gold (I) complexes for anti-cancer treatment. Chem. Commun. 2011, 47, 9554–9560. [Google Scholar] [CrossRef]
- Shaw, C.F. Gold-based therapeutic agents. Chem. Rev. 1999, 99, 2589–2600. [Google Scholar] [CrossRef]
- Gabbiani, C.; Messori, L. Protein targets for anticancer gold compounds: Mechanistic inferences. Anticancer Agents Med. Chem. 2011, 11, 929–939. [Google Scholar] [CrossRef]
- Sun, R.W.Y.; Li, C.K.L.; Ma, D.L.; Yan, J.J.; Lok, C.N.; Leung, C.H.; Zhu, N.; Che, C.M. Stable anticancer gold (III)–porphyrin complexes: Effects of porphyrin structure. Chem. Eur. J. 2010, 16, 3097–3113. [Google Scholar] [CrossRef]
- Casini, A.; Messori, L. Molecular mechanisms and proposed targets for selected anticancer gold compounds. Curr. Top. Med. Chem. 2011, 11, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashmi, A.S.K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Gorin, D.J.; Sherry, B.D.; Toste, F.D. Ligand effects in homogeneous Au catalysis. Chem. Rev. 2008, 108, 3351–3378. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Brouwer, C.; He, C. Gold-catalyzed organic transformations. Chem. Rev. 2008, 108, 3239–3265. [Google Scholar] [CrossRef] [PubMed]
- Marion, N.; Nolan, S.P. N-heterocyclic carbenes in gold catalysis. Chem. Soc. Rev. 2008, 37, 1776–1782. [Google Scholar] [CrossRef]
- Barnard, P.J.; Berners-Price, S.J. Targeting the mitochondrial cell death pathway with gold compounds. Coord. Chem. Rev. 2007, 251, 1889–1902. [Google Scholar] [CrossRef]
- Navarro, M. Gold complexes as potential anti-parasitic agents. Coord. Chem. Rev. 2009, 253, 1619–1626. [Google Scholar] [CrossRef]
- Krishnamurthy, D.; Karver, M.R.; Fiorillo, E.; Orrú, V.; Stanford, S.M.; Bottini, N.; Barrios, M. Gold(I)-mediated inhibition of protein tyrosine phosphatases: A detailed in vitro and cellular study. J. Med. Chem. 2008, 51, 4790–4795. [Google Scholar] [CrossRef]
- Glišić, B.D.; Djuran, M.I. Gold complexes as antimicrobial agents: An overview of different biological activities in relation to the oxidation state of the gold ion and the ligand structure. Dalton Trans. 2014, 43, 5950–5969. [Google Scholar] [CrossRef]
- Liu, W.; Gust, R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L.A.; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W.S.; Wolber, G. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010, 53, 8608–8618. [Google Scholar] [CrossRef] [PubMed]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic carbene metal complexes in medicinal chemistry. Dalton Trans. 2013, 42, 3269–3284. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.; Gaillard, S.; Toye, C.; Macpherson, S.; Nolan, S.P.; Riches, A. Cytotoxicity of gold(I) N-heterocyclic carbene complexes assessed by using human tumor cell lines. Chem. Eur. J. 2011, 17, 6620–6624. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.J.; Ruhayel, R.A.; Barnard, P.J.; Baker, M.V.; Berners-Price, S.J.; Filipovska, A. Mitochondria-targeted chemotherapeutics: The rational design of gold(I) N-heterocyclic carbene complexes that are selectively toxic to cancer cells and target protein selenols in preference to thiols. J. Am. Chem. Soc. 2008, 130, 12570–12571. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; Casini, A. A golden future in medicinal inorganic chemistry: The promise of anticancer gold organometallic compounds. Dalton Trans. 2014, 43, 4209–4219. [Google Scholar] [CrossRef]
- Schuh, E.; Pflüger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold (I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef]
- Arambula, J.F.; McCall, R.; Sidoran, K.J.; Magda, D.; Mitchell, N.A.; Bielawski, C.W.; Lynch, V.M.; Sessler, J.L.; Arumugam, K. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. Chem. Sci. 2016, 7, 1245–1256. [Google Scholar] [CrossRef]
- Pratesi, A.; Gabbiani, C.; Michelucci, E.; Ginanneschi, M.; Papini, A.M.; Rubbiani, R.; Ott, I.; Messori, L. Insights on the mechanism of thioredoxin reductase inhibition by gold N-heterocyclic carbene compounds using the synthetic linear selenocysteine containing C-terminal peptide hTrxR(488–499): An ESI-MS investigation. J. Inorg. Biochem. 2014, 136, 161–169. [Google Scholar] [CrossRef]
- Arcau, J.; Andermark, V.; Rodrigues, M.; Giannicchi, I.; PérezGarcia, L.; Ott, I.; Rodríguez, L. Synthesis and Biological Activity of Gold(I) N-Heterocyclic Carbene Complexes with Long Aliphatic Side Chains. Eur. J. Inorg. Chem. 2014, 35, 6117–6125. [Google Scholar] [CrossRef]
- Urig, S.; Fritz-Wolf, K.; Réau, R.; Herold-Mende, C.; Tóth, K.; Davioud-Charvet, E.; Becker, K. Undressing of Phosphine Gold(I) Complexes as Irreversible Inhibitors of Human Disulfide Reductases. Angew. Chem. Int. Ed. 2006, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Scaletti, F.; Massai, L.; Cinellu, M.A.; Russo Krauss, I.; Di Martino, G.; Vergara, A.; Paduano, L.; Merlino, A. Interactions of gold-based drugs with proteins: Crystal structure of the adduct formed between ribonuclease A and a cytotoxic gold (III) compound. Metallomics 2014, 6, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Scaletti, F.; Massai, L.; Cinellu, M.A.; Gabbiani, C.; Vergara, A.; Merlino, A. The mode of action of anticancer gold-based drugs: A structural perspective. Chem. Commun. 2013, 49, 10100–10102. [Google Scholar]
- Ferraro, G.; Massai, L.; Messori, L.; Cinellu, M.A.; Merlino, A. Structural evidences for a secondary gold binding site in the hydrophobic box of lysozyme. Biometals 2015, 28, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.; Gabbiani, C.; Merlino, A. First crystal structure for a gold carbene–protein adduct. Bioconjugate Chem. 2016, 27, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, C.; Scaletti, F.; Massai, L.; Michelucci, E.; Cinellu, M.A.; Messori, L. Medicinal gold compounds form tight adducts with the copper chaperone Atox-1: Biological and pharmacological implications. Chem. Commun. 2012, 48, 11623–11625. [Google Scholar] [CrossRef] [PubMed]
- Bhabak, K.P.; Bhuyan, B.J.; Mugesh, G. Bioinorganic and medicinal chemistry: Aspects of gold(I)-protein complexes. Dalton Trans. 2011, 40, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Serebryanskaya, T.V.; Lyakhov, A.S.; Ivashkevich, L.S.; Schur, J.; Frias, C.; Prokop, A.; Ott, I. Gold (I) thiotetrazolates as thioredoxin reductase inhibitors and antiproliferative agents. Dalton Trans. 2015, 44, 1161–1169. [Google Scholar] [CrossRef]
- Vergara, E.; Casini, A.; Sorrentino, F.; Zava, O.; Cerrada, E.; Rigobello, M.P.; Bindoli, A.; Laguna, M.; Dyson, P.J. Anticancer therapeutics that target selenoenzymes: Synthesis, characterization, in vitro cytotoxicity, and thioredoxin reductase inhibition of a series of gold(I) complexes containing hydrophilic phosphine ligands. ChemMedChem 2010, 5, 96–102. [Google Scholar] [CrossRef]
- Ott, I.; Qian, X.; Xu, Y.; Vlecken, D.H.W.; Marques, I.J.; Kubutat, D.; Will, J.; Sheldrick, W.S.; Jesse, P.; Prokop, A. A gold(I) phosphine complex containing a naphthalimide ligand functions as a TrxR inhibiting antiproliferative agent and angiogenesis inhibitor. J. Med. Chem. 2009, 52, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.V.; Barnard, P.J.; Berners-Price, S.J.; Brayshaw, S.K.; Hickey, J.L.; Skelton, B.W.; White, A.H. Cationic, linear Au(I) N-heterocyclic carbene complexes: Synthesis, structure and anti-mitochondrial activity. Dalton Trans. 2006, 30, 3708–3715. [Google Scholar] [CrossRef] [PubMed]
- Aher, S.B.; Muskawar, P.N.; Thenmozhi, K.; Bhagat, P.R. Recent developments of metal N-heterocyclic carbenes as anticancer agents. Eur. J. Med. Chem. 2014, 81, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Che, C.M.; Ott, I. Caffeine derived platinum(II) N-heterocyclic carbene complexes with multiple anti-cancer activities. J. Organomet. Chem. 2015, 782, 37–41. [Google Scholar] [CrossRef]
- Wang, H.M.; Vasam, C.S.; Tsai, T.Y.; Chen, S.H.; Chang AH, H.; Lin, I.J. Gold (I) N-heterocyclic carbene and carbazolate complexes. Organometallics 2005, 24, 486–493. [Google Scholar] [CrossRef]
- Messori, L.; Cinellu, M.A.; Merlino, A. Protein recognition of gold-based drugs: 3D structure of the complex formed when lysozyme reacts with Aubipyc. ACS Med. Chem. Lett. 2014, 5, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Marchetti, L.; Massai, L.; Scaletti, F.; Guerri, A.; Landini, I.; Nobili, S.; Perrone, G.; Mini, E.; Leoni, P.; et al. Chemistry and biology of two novel gold (I) carbene complexes as prospective anticancer agents. Inorg. Chem. 2014, 53, 2396–2403. [Google Scholar] [CrossRef]
- Rubbiani, R. Gold (I) N-Heterocyclic Carbene Complexes: A Chemical and Biological Study of Their Therapeutic Potential as New Anticancer Agents. Ph.D. Thesis, TU Braunschweig, Braunschweig, Germany, 2013. [Google Scholar]
- Minenkov, Y.; Singstad, Å.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Qi, S.C.; Hayashi, J.I.; Zhang, L. Recent application of calculations of metal complexes based on density functional theory. RSC Adv. 2016, 6, 77375–77395. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. How accurate are the Minnesota density functionals for noncovalent interactions, isomerization energies, thermochemistry, and barrier heights involving molecules composed of main-group elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef]
- Grigorenko, B.R.; Khrenova, M.G.; Nemukin, A.V. Amide–imide tautomerization in the glutamine side chain in enzymatic and photochemical reactions in proteins. Phys. Chem. Chem. Phys. 2018, 20, 23827–23836. [Google Scholar] [PubMed]
- Zou, J.; Taylor, P.; Dornan, J.; Robinson, S.P.; Walkinshaw, M.D.; Sadler, P.J. First crystal structure of a medicinally relevant gold protein complex: Unexpected binding of [Au(PEt3)]+ to histidine. Angew. Chem. Int. Ed. Engl. 2000, 39, 2931–2934. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef] [Green Version]
- Jaguar Version 7.5; Schrodinger LLC: New York, NY, USA, 2008.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. (Eds.) Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Niu, S.; Hall, B.M. Theoretical studies on reactions of transition-metal complexes. Chem. Rev. 2000, 100, 353–405. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Insight into the electrochemical reduction mechanism of Pt (IV) anticancer complexes. Inorg. Chem. 2018, 57, 3411–3419. [Google Scholar] [CrossRef] [PubMed]
- Paciotti, R.; Corinti, D.; De Petris, A.; Ciavardini, A.; Piccirillo, S.; Coletti, C.; Re, N.; Maitre, P.; Bellina, B.; Barran, P.; et al. Cisplatin and transplatin interaction with methionine: Bonding motifs assayed by vibrational spectroscopy in the isolated ionic complexes. Phys. Chem. Chem. Phys. 2017, 19, 26697–26707. [Google Scholar] [PubMed]
- Corinti, D.; De Petris, A.; Coletti, C.; Re, N.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Cisplatin primary complex with L-histidine target revealed by IR multiple photon dissociation (IRMPD) spectroscopy. Chem. Phys. Chem. 2017, 18, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Corinti, D.; Coletti, C.; Re, N.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Cisplatin binding to biological ligands revealed at the encounter complex level by IR action spectroscopy. Chem. Eur. J. 2016, 22, 3794–3803. [Google Scholar] [CrossRef] [PubMed]
- Graziani, V.; Coletti, C.; Marrone, A.; Re, N. Activation and reactivity of a bispidine analogue of cisplatin: A theoretical investigation. J. Phys. Chem. A 2016, 120, 5175–5186. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Coletti, C.; Marrone, A.; Re, N. Activation of carboplatin by carbonate: A theoretical investigation. Dalton Trans. 2012, 41, 12960–12969. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Coletti, C.; Marrone, A.; Re, N. Activation of carboplatin by chloride ions: A theoretical investigation. Theor. Chem. Acc. 2011, 129, 757–769. [Google Scholar] [CrossRef]
- Besker, N.; Coletti, C.; Marrone, A.; Re, N. Binding of antitumor ruthenium complexes to DNA and proteins: A theoretical approach. J. Phys. Chem. B 2007, 111, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Paciotti, R.; Tolbatov, I.; Graziani, V.; Marrone, A.; Re, N.; Coletti, C. Insights on the Activity of Platinum-based Anticancer Complexes through Computational Methods. AIP Conf. Proc. 2018, 2040, 020019. [Google Scholar]
- Marino, T.; Russo, N.; Toscano, M.; Pavelka, M. Theoretical investigation on DNA/RNA base pairs mediated by copper, silver, and gold cations. Dalton Trans. 2012, 41, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Nuevo, D.; Poyatos, M.; Peris, E. A dinuclear Au(I) complex with a pyrene-di-N-heterocyclic carbene linker: Supramolecular and catalytic studies. Organometallics 2018, 37, 3407–3411. [Google Scholar] [CrossRef]
- Graziani, V.; Marrone, A.; Re, N.; Coletti, C.; Platts, J.A.; Casini, A. A multi-level theoretical study to disclose the binding mechanisms of gold (III)–bipyridyl compounds as selective aquaglyceroporin inhibitors. Chem. Eur. J. 2017, 23, 13802–13813. [Google Scholar] [CrossRef]
- Wenzel, M.; Mósca, A.; Graziani, V.; Aikman, B.; Thomas, S.; de Almeida, A.; Platts, J.; Re, N.; Coletti, C.; Marrone, A.; et al. Insights into the Mechanisms of Aquaporin-3 Inhibition by Gold(III) Complexes: The Importance of Non-Coordinative Adduct Formation. Inorg. Chem. 2019, 58, 2140–2148. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular orbital methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z= 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.B.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Neto, A.C.; Muniz, E.P.; Centoducatte, R.; Jorge, F.E. Gaussian basis sets for correlated wave functions. Hydrogen, helium, first-and second-row atoms. J. Mol. Struct. 2005, 718, 219–224. [Google Scholar] [CrossRef]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Ringnalda, M.; Goddard, W.A., III; Honig, B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New model for calculation of solvation free energies: Correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
- Wertz, D.H. Relationship between the gas-phase entropies of molecules and their entropies of solvation in water and 1-octanol. J. Am. Chem. Soc. 1980, 102, 5316–5322. [Google Scholar] [CrossRef]
- Cooper, J.; Ziegler, T. A density functional study of SN2 substitution at square-planar platinum (II) complexes. Inorg. Chem. 2002, 41, 6614–6622. [Google Scholar] [CrossRef]
- Lau, J.K.C.; Deubel, D.V. Hydrolysis of the anticancer drug cisplatin: Pitfalls in the interpretation of quantum chemical calculations. J. Chem. Theory Comput. 2006, 2, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, M.; Nielsen, R.J.; Periana, R.A.; Goddard, W.A., III. Product protection, the key to developing high performance methane selective oxidation catalysts. J. Am. Chem. Soc. 2009, 131, 17110–17115. [Google Scholar] [CrossRef]
- Zhu, H.; Ziegler, T. Probing the influence of trans and leaving ligands on the ability of square-planar platinum (II) complexes to activate methane. A theoretical study. Organometallics 2009, 28, 2773–2777. [Google Scholar] [CrossRef]
- Bercaw, J.E.; Chen, G.S.; Labinger, J.A.; Lin, B.L. Protonolysis of platinum (II) and palladium (II) methyl complexes: A combined experimental and theoretical investigation. Organometallics 2010, 29, 4354–4359. [Google Scholar] [CrossRef]
- Ben-Naim, A.Y. Solvation Thermodynamics; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 1995, 91, 43–56. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
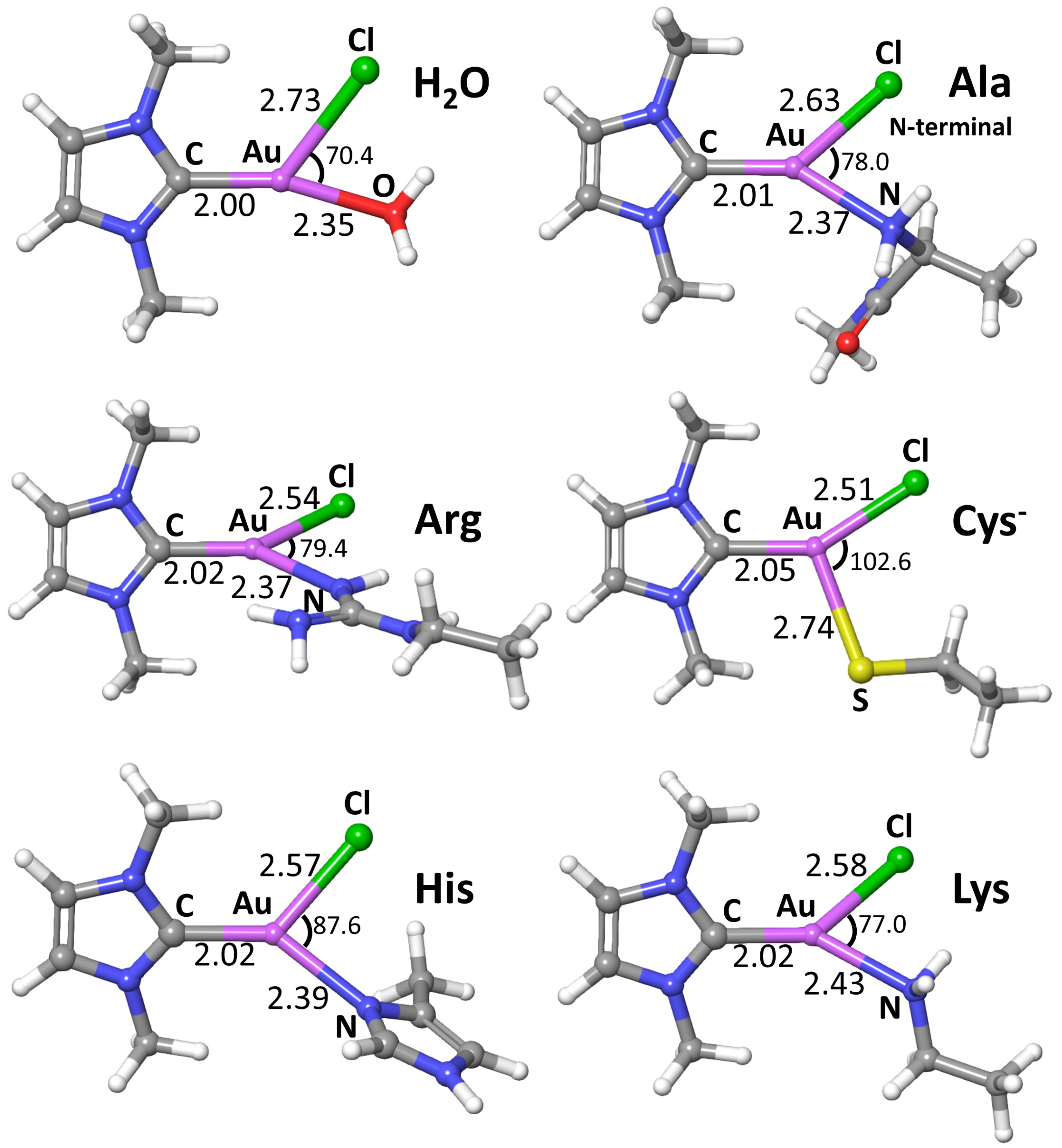
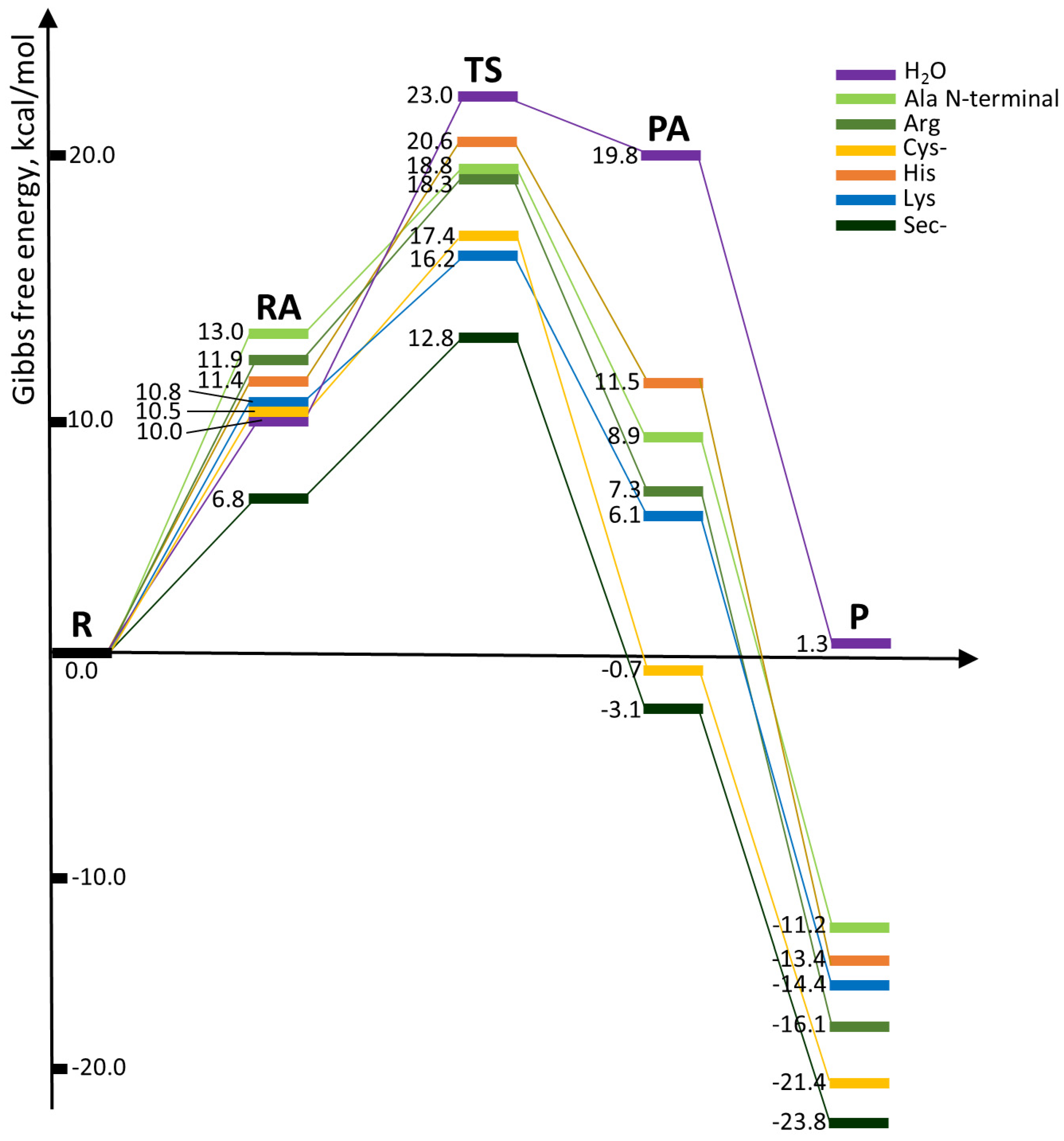
| Reaction with | R → P | R → TS | ||
|---|---|---|---|---|
| ΔH | ΔG | ΔH | ΔG | |
| H2O | 7.2 | 1.3 | 16.8 | 23.0 |
| Ala N-terminal | −6.6 | −11.2 | 11.9 | 18.8 |
| Arg | −11.0 | −16.1 | 11.3 | 18.3 |
| Cys | −3.5 | −9.1 | 10.9 | 18.1 |
| Cys– | −16.8 | −21.4 | 11.6 | 17.4 |
| His | −8.3 | −13.4 | 14.1 | 20.6 |
| Lys | −10.2 | −14.4 | 9.0 | 16.2 |
| Met | −5.4 | −9.8 | 11.5 | 18.2 |
| Sec | −2.2 | −6.7 | 11.2 | 17.6 |
| Sec– | −19.2 | −23.8 | 6.7 | 12.8 |
| Gln/Asn (amidic form) | 6.0 | 2.4 | 17.5 | 24.8 |
| Gln/Asn (imidic form) * | −7.3 | −12.6 | 10.0 | 16.8 |
| Glu/Asp acid | −2.2 | −7.0 | 18.2 | 24.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Reactivity of Gold(I) Monocarbene Complexes with Protein Targets: A Theoretical Study. Int. J. Mol. Sci. 2019, 20, 820. https://doi.org/10.3390/ijms20040820
Tolbatov I, Coletti C, Marrone A, Re N. Reactivity of Gold(I) Monocarbene Complexes with Protein Targets: A Theoretical Study. International Journal of Molecular Sciences. 2019; 20(4):820. https://doi.org/10.3390/ijms20040820
Chicago/Turabian StyleTolbatov, Iogann, Cecilia Coletti, Alessandro Marrone, and Nazzareno Re. 2019. "Reactivity of Gold(I) Monocarbene Complexes with Protein Targets: A Theoretical Study" International Journal of Molecular Sciences 20, no. 4: 820. https://doi.org/10.3390/ijms20040820
APA StyleTolbatov, I., Coletti, C., Marrone, A., & Re, N. (2019). Reactivity of Gold(I) Monocarbene Complexes with Protein Targets: A Theoretical Study. International Journal of Molecular Sciences, 20(4), 820. https://doi.org/10.3390/ijms20040820