Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury


Abstract
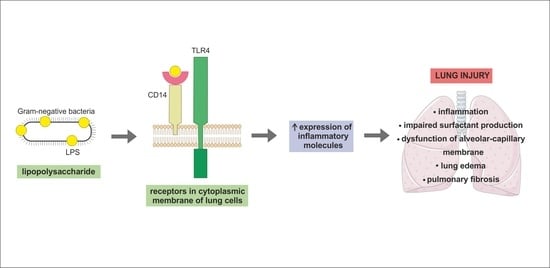
:
{kind=link}
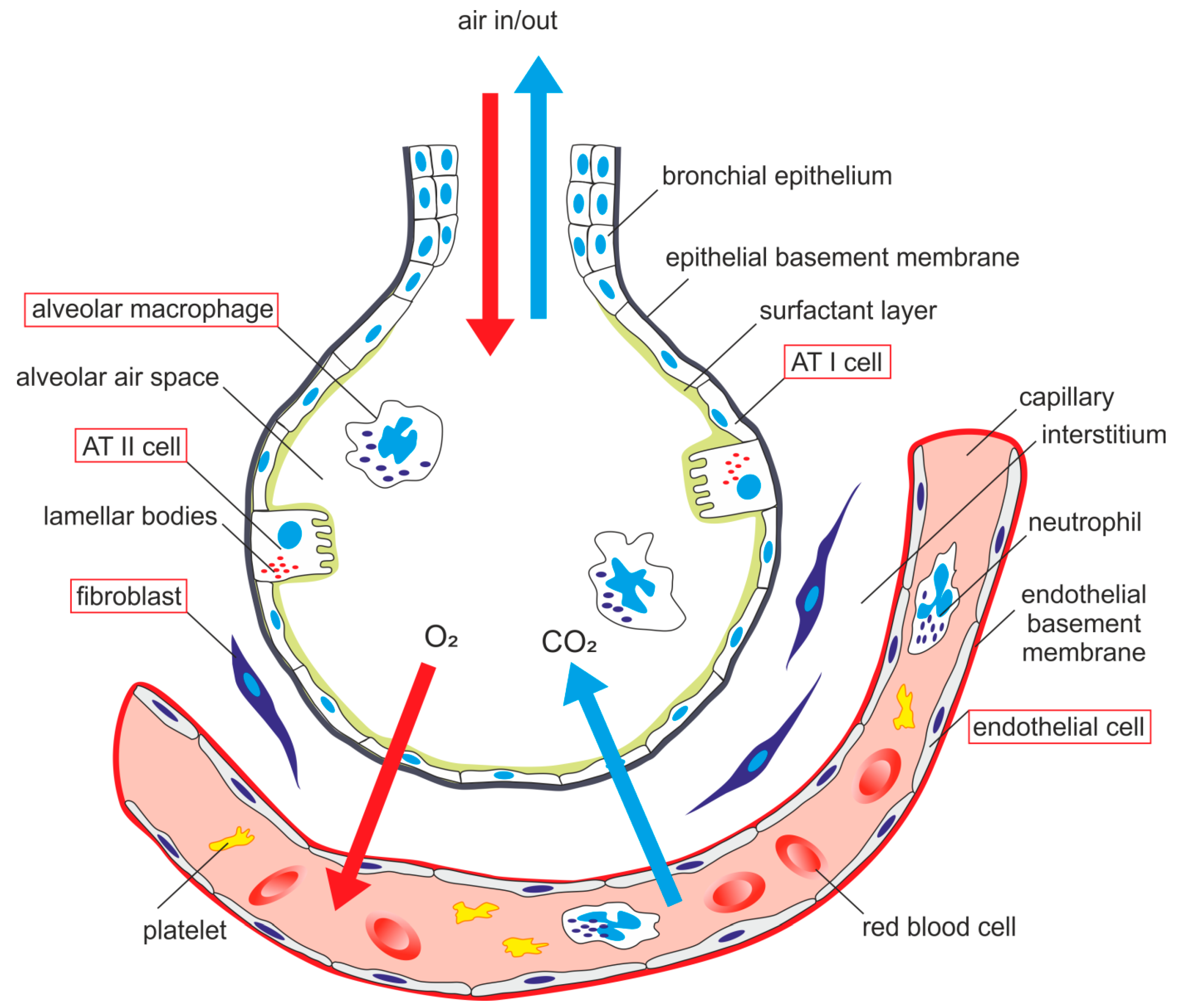{kind=link}
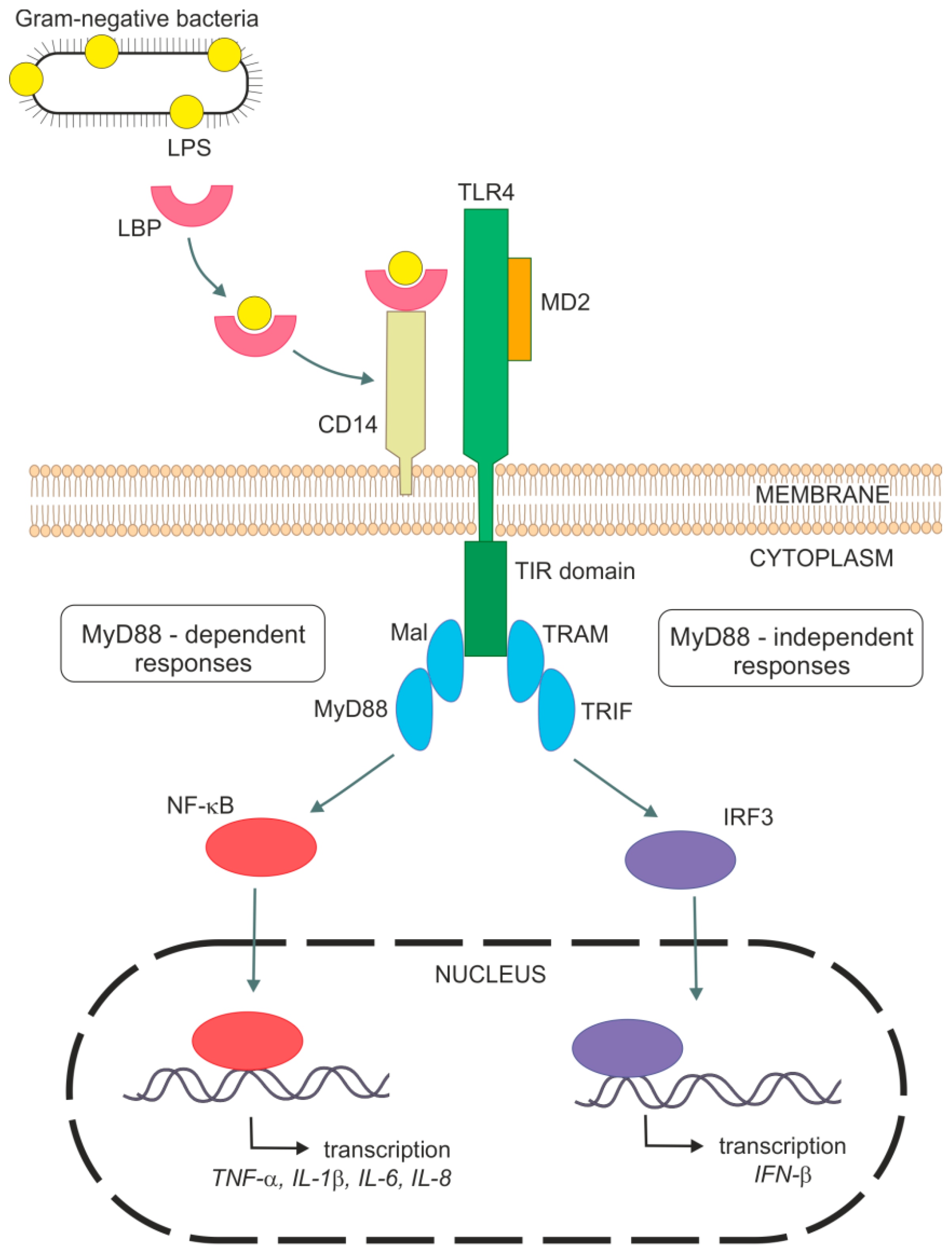{kind=link}
1. Introduction
2. Mechanism of LPS Signal Transduction Pathway in Lung Cells
3. Alveolar Epithelial Type I Cells
Interaction of ATI Cells with LPS
4. Alveolar Epithelial Type II Cells
4.1. Interaction of ATII Cells with LPS
4.2. Interaction of Pulmonary Surfactant with LPS
5. Endothelial Cells
Interaction of Endothelial Cells with LPS
6. Alveolar Macrophages
Interaction of Alveolar Macrophages with LPS
7. Lung Fibroblasts
Interaction of Lung Fibroblasts with LPS
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALI | acute lung injury |
| AM | alveolar macrophages |
| ARDS | acute respiratory distress syndrome |
| ATI cells | alveolar epithelial type I cells |
| ATII cells | alveolar epithelial type II cells |
| COPD | chronic obstructive pulmonary disease |
| DPPC | dipalmitoylphosphatidylcholine |
| ICAM-1 | intercellular Adhesion Molecule 1 |
| IFN- β | interferon β |
| IFN- γ | interferon γ |
| IL | interleukin |
| IL-1R | interleukin-1 receptor |
| IPF | idiopathic pulmonary fibrosis |
| LBP | LPS-binding protein |
| Mal | MyD88 adaptor-like protein |
| mCD14 | membrane-bound CD14 |
| MCP-1 | monocyte chemoattractant protein 1 |
| MD2 | myeloid differentiation protein 2 |
| MIP-3α | macrophage inflammatory protein 3α |
| MyD88 | myeloid differentiation factor 88 |
| NF- κB | nuclear factor-κB |
| NO | nitric oxide |
| PAMP | pathogen-associated molecular pattern |
| PPRs | pattern-recognition receptors |
| RAGE | receptor for advanced glycation end products |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| sCD14 | Soluble CD14 |
| SIRPα | Signal regulatory protein α |
| SP-A | surfactant protein A |
| SP-B | surfactant protein B |
| SP-C | surfactant protein C |
| SP-D | surfactant protein D |
| SPs | surfactant proteins |
| sRAGE | soluble isoform of receptor for advanced glycation end products |
| TGF-β | transforming growth factor β |
| TIR domains | Toll/IL-1R homology domains |
| TLRs | toll-like receptors |
| TNF- α | tumor necrosis factor α |
| TRAM | TRIF-related adaptor molecule |
| TRIF | TIR-domain-containing adaptor protein |
| VCAM-1 | vascular cell adhesion molecule 1 |
References
- Guillot, L.; Nathan, N.; Tabary, O.; Thouvenin, G.; Le Rouzic, P.; Corvol, H.; Amselem, S.; Clement, A. Alveolar Epithelial Cells: Master Regulators of Lung Homeostasis. Int. J. Biochem. Cell Biol. 2013, 45, 2568–2573. [Google Scholar] [CrossRef] [PubMed]
- Losa, D.; Chanson, M. The Lung Communication Network. Cell. Mol. Life Sci. 2015, 72, 2793–2808. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. Lung Morphometry: The Link between Structure and Function. Cell Tissue Res. 2017, 367, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Meredith, T.C.; Kahne, D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomaznik, M.; Nova, Z.; Calkovska, A. Pulmonary Surfactant and Bacterial Lipopolysaccharide: The Interaction and its Functional Consequences. Physiol. Res. 2017, 66, S147–S157. [Google Scholar] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef]
- Płóciennikowska, A.; Hromada-Judycka, A.; Borzęcka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef]
- Chow, J.C.; Young, D.W.; Golenbock, D.T.; Christ, W.J.; Gusovsky, F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 1999, 274, 10689–10692. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Young, D.W.; Gusovsky, F.; Chow, J.C. Cellular events mediated by lipopolysaccharide-stimulated toll-like receptor 4. MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J. Biol. Chem. 2000, 275, 20861–20866. [Google Scholar] [CrossRef] [PubMed]
- Nativel, B.; Couret, D.; Giraud, P.; Meilhac, O.; d’Hellencourt, C.L.; Viranaïcken, W.; Da Silva, C.R. Porphyromonas gingivalis lipopolysaccharides act exclusively through TLR4 with a resilience between mouse and human. Sci. Rep. 2017, 7, 15789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Takeuchi, O.; Hoshino, K.; Kawai, T.; Sanjo, H.; Takada, H.; Ogawa, T.; Takeda, K.; Akira, S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 1999, 11, 443–451. [Google Scholar] [CrossRef]
- Wang, P.; Han, X.; Mo, B.; Huang, G.; Wang, C. LPS enhances TLR4 expression and IFN-γ production via the TLR4/IRAK/NF-κB signaling pathway in rat pulmonary arterial smooth muscle cells. Mol. Med. Rep. 2017, 16, 3111–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Arbour, N.C.; Lorenz, E.; Schutte, B.C.; Zabner, J.; Kline, J.N.; Jones, M.; Frees, K.; Watt, J.L.; Schwartz, D.A. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 2000, 25, 187–191. [Google Scholar] [CrossRef]
- Poltorak, A.; Ricciardi-Castagnoli, P.; Citterio, S.; Beutler, B. Physical contact between LPS and Tlr4 revealed by genetic complementation. Proc. Natl. Acad. Sci. USA 2000, 97, 2163–2167. [Google Scholar] [CrossRef]
- Lien, E.; Means, T.K.; Heine, H.; Yoshimura, A.; Kusumoto, S.; Fukase, K.; Fenton, M.J.; Oikawa, M.; Qureshi, N.; Monks, B.; et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin Investig. 2000, 105, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Correia, J.; Soldau, K.; Christen, U.; Tobias, P.S.; Ulevitch, R.J. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex: Transfer from CD14 to TLR4 and MD-2. J. Biol. Chem. 2001, 276, 21129–21135. [Google Scholar] [CrossRef] [PubMed]
- Schumann, R.R.; Leong, S.R.; Flaggs, G.W.; Gray, P.W.; Wright, S.D.; Mathison, J.C.; Tobias, P.S.; Ulevitch, R.J. Structure and function of lipopolysaccharide binding protein. Science 1990, 249, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, M.; Muroi, M.; Tanamoto, K.; Suzuki, T.; Azuma, H.; Ikeda, H. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: Roles of the receptor complex. Pharmacol. Ther. 2003, 100, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.M. Dynamic lipopolysaccharide transfer cascade to TLR4/MD2 complex via LBP and CD14. BMB Rep. 2017, 50, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugin, J.; Heumann, I.D.; Tomasz, A.; Kravchenko, V.V.; Akamatsu, Y.; Nishijima, M.; Glauser, M.P.; Tobias, P.S.; Ulevitch, R.J. CD14 is a pattern recognition receptor. Immunity 1994, 1, 509–516. [Google Scholar] [CrossRef]
- Tapping, R.I.; Tobias, P.S. Soluble CD14-mediated cellular responses to lipopolysaccharide. Chem. Immunol. 2000, 74, 108–121. [Google Scholar] [PubMed]
- Zanoni, I.; Granucci, F. Role of CD14 in host protection against infections and in metabolism regulation. Front. Cell. Infect. Microbiol. 2013, 3, 32. [Google Scholar] [CrossRef]
- Haziot, A.; Ferrero, E.; Köntgen, F.; Hijiya, N.; Yamamoto, S.; Silver, J.; Stewart, C.L.; Goyert, S.M. Resistance to endotoxin shock and reduced dissemination of Gram-negative bacteria in CD14-deficient mice. Immunity 1996, 4, 407–414. [Google Scholar] [CrossRef]
- Gioannini, T.L.; Teghanemt, A.; Zhang, D.; Coussens, N.P.; Dockstader, W.; Ramaswamy, S.; Weiss, J.P. Isolation of an endotoxin–MD 2 complex that produces Toll like receptor 4 dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. USA 2004, 101, 4186–4191. [Google Scholar] [CrossRef]
- Haziot, A.; Lin, X.Y.; Zhang, F.; Goyert, S.M. The induction of acute phase proteins by lipopolysaccharide uses a novel pathway that is CD14-independent. J. Immunol. 1998, 160, 2570–2572. [Google Scholar]
- Kimura, S.; Tamamura, T.; Nakagawa, I.; Koga, T.; Fujiwara, T.; Hamada, S. CD14-dependent and independent pathways in lipopolysaccharide-induced activation of a murine B-cell line, CH12. LX. Scand. J. Immunol. 2000, 51, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kumazawa, Y.; Inoue, J. Liposomal lipopolysaccharide initiates TRIF-dependent signaling pathway independent of CD14. PLoS ONE 2013, 8, e60078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Huber, M.; Kalis, C.; Keck, S.; Galanos, C.; et al. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005, 6, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Kalis, C.; Keck, S.; Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Beutler, B.; et al. R-form LPS, the master key to the activation ofTLR4/MD-2-positive cells. Eur. J. Immunol. 2006, 36, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, B.; Rietschel, E. Innate immune sensing and its roots: The story of endotoxin. Nat. Rev. Immunol. 2003, 3, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Akashi, S.; Nagafuku, M.; Ogata, M.; Iwakura, Y.; Akira, S.; Kitamura, T.; Kosugi, A.; Kimoto, M.; Miyake, K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 2002, 3, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Schromm, A.B.; Lien, E.; Henneke, P.; Chow, J.C.; Yoshimura, A.; Heine, H.; Latz, E.; Monks, B.G.; Schwartz, D.A.; Miyake, K.; et al. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: A point mutation in a conserved region of MD-2 abolishes endotoxin-induced signalling. J. Exp. Med. 2001, 194, 79–88. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR domain containing adaptors in Toll like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Feng, C.G.; Scanga, C.A.; Collazo-Custodio, C.M.; Cheever, A.W.; Hieny, S.; Caspar, P.; Sher, A. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J. Immunol. 2003, 171, 4758–4764. [Google Scholar] [CrossRef]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Sasai, M.; Shida, K.; Fujita, T.; Matsumoto, M.; Seya, T. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-beta. J. Biol. Chem. 2003, 278, 49751–49762. [Google Scholar] [CrossRef] [PubMed]
- Pålsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zughaier, S.M.; Zimmer, S.M.; Datta, A.; Carlson, R.W.; Stephens, D.S. Differential Induction of the Toll-Like Receptor 4-MyD88-Dependent and -Independent Signaling Pathways by Endotoxins. Infect. Immun. 2005, 73, 2940–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieser, K.J.; Kagan, J.C. Multi-receptor detection of individual bacterial products by the innate immune system. Nat. Rev. Immunol. 2017, 17, 376–390. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Fonceca, A.M.; Zosky, G.R.; Bozanich, E.M.; Sutanto, E.N.; Kicic, A.; McNamara, P.S.; Knight, D.A.; Sly, P.D.; Turner, D.J.; Stick, S.M. Accumulation mode particles and LPS exposure induce TLR-4 dependent and independent inflammatory responses in the lung. Respir. Res. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denlinger, L.C.; Fisette, P.L.; Sommer, J.A.; Watters, J.J.; Prabhu, U.; Dubyak, G.R.; Proctor, R.A.; Bertics, P.J. Cutting edge: The nucleotide receptor P2X7 contains multiple protein- and lipid-interaction motifs including a potential binding site for bacterial lipopolysaccharide. J. Immunol. 2001, 167, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Monção-Ribeiro, L.C.; Cagido, V.R.; Lima-Murad, G.; Santana, P.T.; Riva, D.R.; Borojevic, R.; Zin, W.A.; Cavalcante, M.C.; Riça, I.; Brando-Lima, A.C.; et al. Lipopolysaccharide-induced lung injury: Role of P2X7 receptor. Respir. Physiol. Neurobiol. 2011, 179, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Volonté, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2X7 receptors: Channels, pores and more. CNS Neurol. Disord. Drug Targets 2012, 11, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Costa-Junior, H.M.; Sarmento Vieira, F.; Coutinho-Silva, R. C terminus of the P2X7 receptor: Treasure hunting. Purinergic Signal. 2011, 7, 7–19. [Google Scholar] [CrossRef]
- Dagvadorj, J.; Shimada, K.; Chen, S.; Jones, H.D.; Tumurkhuu, G.; Zhang, W.; Wawrowsky, K.A.; Crother, T.R.; Arditi, M. Lipopolysaccharide Induces Alveolar Macrophage Necrosis via CD14 and the P2X7 Receptor Leading to Interleukin-1α Release. Immunity 2015, 42, 640–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J. Biology of Alveolar Type II Cells. Respirology 2006, 11, S12–S15. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, E.; Perez-Gil, J. Structure-function relationships in pulmonary surfactant membranes: From biophysics to therapy. Biochim. Biophys. Acta 2014, 1838, 1568–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, M.F.; Moodley, Y. When Is an Alveolar Type 2 Cell an Alveolar Type 2 Cell? A Conundrum for Lung Stem Cell Biology and Regenerative Medicine. Am. J. Respir. Cell Mol. Biol. 2017, 57, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.L.; Brody, A.R.; Colby, T.V.; Mason, R.; Williams, M.C. Knowns and unknowns of the alveolus. Proc. Am. Thorac. Soc. 2008, 5, 778–782. [Google Scholar] [CrossRef]
- Griffiths, M.J.; Bonnet, D.; Janes, S.M. Stem Cells of the Alveolar epithelium. Lancet 2005, 366, 249–260. [Google Scholar] [CrossRef]
- Williams, M.C. Alveolar Type I Cells: Molecular Phenotype and Development. Annu. Rev. Physiol. 2003, 65, 669–695. [Google Scholar] [CrossRef]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.J.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx(+) Type I Alveolar Cells to Regenerate Type II Cells in the Lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef]
- Johnson, M.D.; Widdicombe, J.H.; Allen, L.; Barbry, P.; Dobbs, L.G. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc. Natl. Acad. Sci. USA 2002, 99, 1966–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toczylowska-Maminska, R.; Dolowy, K. Ion transporting proteins of human bronchial epithelium. J. Cell. Biochem. 2012, 113, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.; Johnson, M.D. Differential response of primary alveolar type I and type II cells to LPS stimulation. PLoS ONE 2013, 8, e55545. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ferrari, J.D.; Cao, Y.; Ramirez, M.I.; Jones, M.R.; Quinton, L.J.; Mizgerd, J.P. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 2012, 189, 2450–2459. [Google Scholar] [CrossRef] [PubMed]
- Nova, Z.; Mokra, D.; Vidomanova, E.; Kolomaznik, M.; Skovierova, H.; Halasova, E.; Calkovska, A. Effect of lipopolysaccharide on alveolar epithelial type II cells. Acta Physiol. 2017, 221 (Suppl. S713), 238. [Google Scholar]
- Li, Y.; Wu, R.; Zhao, S.; Cheng, H.; Ji, P.; Yu, M.; Tian, Z. RAGE/NF-κB pathway mediates lipopolysaccharide-induced inflammation in alveolar type I epithelial cells isolated from neonate rats. Inflammation 2014, 37, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Shirasawa, M.; Ware, L.B.; Kojima, K.; Hata, Y.; Makita, K.; Mednick, G.; Matthay, Z.A.; Matthay, M.A. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am. J. Respir. Crit. Care Med. 2006, 173, 1008–1015. [Google Scholar] [CrossRef]
- Wong, M.H.; Chapin, O.C.; Johnson, M.D. LPS-stimulated cytokine production in type I cells is modulated by the renin-angiotensin system. Am. J. Respir. Cell Mol. Biol. 2012, 46, 641–650. [Google Scholar] [CrossRef]
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68. [Google Scholar] [CrossRef]
- Kuroki, Y.; Takahashi, M.; Nishitani, C. Pulmonary collectins in innate imunity of the lung. Cell. Microbiol. 2007, 9, 1871–1879. [Google Scholar] [CrossRef]
- Uhliarova, B.; Kopincova, J.; Adamkov, M.; Svec, M.; Calkovska, A. Surfactant proteins A and D are related to severity of the disease, pathogenic bacteria and comorbidity in patients with chronic rhinosinusitis with and without nasal polyps. Clin. Otolaryngol. 2016, 41, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Beers, M.F. Surfactant protein C: Its unique properties and emerging immunomodulatory role in the lung. Microbes Infect. 2006, 8, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Zissel, G.; Ernst, M.; Rabe, K.; Papadopoulos, T.; Magnussen, H.; Schlaak, M.; Muller-Quernheim, J. Human alveolar epithelial cells type II are capable of regulating T-cell activity. J. Investig. Med. 2000, 48, 66–75. [Google Scholar] [PubMed]
- Hiemstra, P.S.; Amatngalim, G.D.; van der Does, A.M.; Taube, C. Antimicrobial Peptides and Innate Lung Defenses: Role in Infectious and Noninfectious Lung Diseases and Therapeutic Applications. Chest 2016, 149, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Mollar, A.; Gay-Jordi, G.; Guillamat-Prats, R.; Closa, D.; Hernandez-Gonzalez, F.; Marin, P.; Burgos, F.; Martorell, J.; Sanchez, M.; Arguis, P.; et al. Safety and Tolerability of Alveolar Type II Cell Transplantation in Idiopathic Pulmonary Fibrosis. Chest 2016, 150, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaher, T.E.; Miller, E.J.; Morrow, D.M.; Javdan, M.; Mantell, L.L. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic. Biol. Med. 2007, 42, 897–908. [Google Scholar] [CrossRef] [PubMed]
- De Prost, N.; Dreyfuss, D. How to prevent ventilator-induced lung injury? Minerva Anestesiol. 2012, 78, 1054–1066. [Google Scholar] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef]
- Cooper, J.R.; Abdullatif, M.B.; Burnett, E.C.; Kempsell, K.E.; Conforti, F.; Tolley, H.; Collins, J.E.; Davies, D.E. Long Term Culture of the A549 Cancer Cell Line Promotes Multilamellar Body Formation and Differentiation towards an Alveolar Type II Pneumocyte Phenotype. PLoS ONE 2016, 11, e0164438. [Google Scholar] [CrossRef]
- Swain, R.J.; Kemp, S.J.; Goldstraw, P.; Tetley, T.D.; Stevens, M.M. Assessment of Cell Line Models of Primary Human Cells by Raman Spectral Phenotyping. Biophys. J. 2010, 98, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- Corbière, V.; Dirix, V.; Norrenberg, S.; Cappello, M.; Remmelink, M.; Mascart, F. Phenotypic characteristics of human type II alveolar epithelial cells suitable for antigen presentation to T lymphocytes. Respir. Res. 2011, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Wu, S.; Li, J.; Fu, W.; He, W.; Liu, X.; Slutsky, A.S.; Zhang, H.; Li, Y. Human alveolar epithelial type II cells in primary culture. Physiol. Rep. 2015, 3, e12288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, C.Y.; Chen, T.L.; Chen, R.M. Molecular mechanisms of lipopolysaccharide-caused induction of surfactant protein-A gene expression in human alveolar epithelial A549 cells. Toxicol. Lett. 2009, 191, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Rucka, Z.; Vanhara, P.; Koutna, I.; Tesarova, L.; Potesilova, M.; Stejskal, S.; Simara, P.; Dolezel, J.; Zvonicek, V.; Coufal, O.; et al. Differential effects of insulin and dexamethasone on pulmonary surfactant-associated genes and proteins in A549 and H441 cells and lung tissue. Int. J. Mol. Med. 2013, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, G.; Müller, G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J. Lipid Res. 1991, 32, 1539–1570. [Google Scholar] [PubMed]
- Armstrong, L.; Medford, A.R.; Uppington, K.M.; Robertson, J.; Witherden, I.R.; Tetley, T.D.; Millar, A.B. Expression of functional toll-like receptor-2 and -4 on alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 241–245. [Google Scholar] [CrossRef]
- Guillot, L.; Medjane, S.; Le-Barillec, K.; Balloy, V.; Danel, C.; Chignard, M.; Si-Tahar, M. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: Evidence for an intracellular compartmentalization of TLR4. J. Biol. Chem. 2004, 279, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Sender, V.; Stamme, C. Lung cell-specific modulation of LPS-induced TLR4 receptor and adaptor localization. Commun. Integr. Biol. 2014, 7, e29053. [Google Scholar] [CrossRef] [PubMed]
- Thorley, A.J.; Grandolfo, D.; Lim, E.; Goldstraw, P.; Young, A.; Tetley, T.D. Innate immune responses to bacterial ligands in the peripheral human lung—Role of alveolar epithelial TLR expression and signalling. PLoS ONE 2011, 6, e21827. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef]
- Pugin, J.; Schürer-Maly, C.C.; Leturcq, D.; Moriarty, A.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc. Natl. Acad. Sci. USA 1993, 90, 2744–2748. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Farkas, L.; Wolf, K.; Kratzel, K.; Eissner, G.; Pfeifer, M. Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549). Scand. J. Immunol. 2002, 56, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Von Schéele, I.; Larsson, K.; Palmberg, L. Interactions between alveolar epithelial cells and neutrophils under pro-inflammatory conditions. Eur. Clin. Respir. J. 2014, 1. [Google Scholar] [CrossRef] [PubMed]
- George, C.L.; White, M.L.; O’Neill, M.E.; Thorne, P.S.; Schwartz, D.A.; Snyder, J.M. Altered surfactant protein A gene expression and protein metabolism associated with repeat exposure to inhaled endotoxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.N.; Zhou, J.H.; Wang, P.; Zhang, X.J. The localization of SP-B and influences of lipopolysaccharide on it. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2338–2345. [Google Scholar] [PubMed]
- Ingenito, E.P.; Mora, R.; Cullivan, M.; Marzan, Y.; Haley, K.; Mark, L.; Sonna, L.A. Decreased surfactant protein-B expression and surfactant dysfunction in a murine model of acute lung injury. Am. J. Respir. Cell Mol. Biol. 2001, 25, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tian, J.; Wang, L.; Wu, W.; Li, H.; Wang, X.; Zeng, X.; Zhang, W. Apoptosis and surfactant protein-C expression inhibition induced by lipopolysaccharide in AEC II cell may associate with NF-κB pathway. J. Toxicol. Sci. 2017, 42, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomaznik, M.; Nova, Z.; Mokra, D.; Zila, I.; Kopincova, J.; Vidomanova, E.; Skovierova, H.; Halasova, E.; Calkovska, A. Modified porcine surfactant restores lung homeostasis in LPS-challenged and artificially ventilated adult rats. Neonatology 2018, 13, 419. [Google Scholar]
- Chuang, C.Y.; Chen, T.L.; Cherng, Y.G.; Tai, Y.T.; Chen, T.G.; Chen, R.M. Lipopolysaccharide induces apoptotic insults to human alveolar epithelial A549 cells through reactive oxygen species-mediated activation of an intrinsic mitochondrion-dependent pathway. Arch. Toxicol. 2011, 85, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Hashimoto, S.; Mizuta, N.; Kobayashi, A.; Kooguchi, K.; Fujiwara, I.; Nakajima, H. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am. J. Respir. Crit. Care Med. 2001, 163, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Vernooy, J.H.; Dentener, M.A.; van Suylen, R.J.; Buurman, W.A.; Wouters, E.F. Intratracheal instillation of lipopolysaccharide in mice induces apoptosis in bronchial epithelial cells: No role for tumor necrosis factor-alpha and infiltrating neutrophils. Am. J. Respir. Cell Mol. Biol. 2001, 24, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.S.; Mura, M.; Seth, R.; Liu, M. Acute lung injury and cell death: How many ways can cells die? Am. J. Physiol. 2008, 294, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, L.; Yu, L.; Han, L.; Ji, W.; Shen, H.; Hu, Z. Time-dependent changes of autophagy and apoptosis in lipopolysaccharide-induced rat acute lung injury. Iran. J. Basic Med. Sci. 2016, 19, 632–637. [Google Scholar] [PubMed]
- Kucukgul, A.; Eedogan, S. Low concentration of oleic acid exacerbates LPS-induced cell death and inflammation in human alveolar epithelial cells. Exp. Lung Res. 2017, 43, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.M.; Chen, T.L.; Lin, Y.L.; Chen, T.G.; Tai, Y.T. Ketamine reduces nitric oxide biosynthesis in human umbilical vein endothelial cells by down-regulating endothelial nitric oxide synthase expression and intracellular calcium levels. Crit. Care Med. 2005, 33, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Tai, Y.T.; Lin, Y.L.; Chen, R.M. Molecular mechanisms of propofol-involved suppression of no biosynthesis and inducible iNOS gene expression in LPS-stimulated macrophage-like raw 264.7 cells. Shock 2010, 33, 93–100. [Google Scholar] [CrossRef] [PubMed]
- García-Verdugo, I.; Cañadas, O.; Taneva, S.G.; Keough, K.M.; Casals, C. Surfactant protein A forms extensive lattice-like structures on 1,2-dipalmitoylphosphatidylcholine/rough-lipopolysaccharide-mixed monolayers. Biophys. J. 2007, 93, 3529–3540. [Google Scholar] [CrossRef]
- Cañadas, O.; Keough, K.M.; Casals, C. Bacterial lipopolysaccharide promotes destabilization of lung surfactant-like films. Biophys. J. 2011, 100, 108–116. [Google Scholar] [CrossRef]
- Kishore, U.; Greenhough, T.J.; Waters, P.; Shrive, A.K.; Ghai, R.; Kamran, M.F.; Bernal, A.L.; Reid, K.B.M.; Madan, T.; Chakraborty, T. Surfactant proteins SP-A and SP-D: Structure, function and receptors. Mol. Immunol. 2006, 43, 1293–1315. [Google Scholar] [CrossRef]
- Gardai, S.J.; Xiao, Y.Q.; Dickinson, M.; Nick, J.A.; Voelker, D.R.; Greene, K.E.; Henson, P.M. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003, 115, 13–23. [Google Scholar] [CrossRef]
- Chaby, R.; Garcia-Verdugo, I.; Espinassous, Q.; Augusto, L.A. Interactions between LPS and lung surfactant proteins. J. Endotoxin Res. 2005, 11, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Van Iwaarden, J.F.; Pikaar, J.C.; van Strijp, J.A. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem. J. 1994, 303, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, H.; Chiba, H.; Iwaki, D.; Sohma, H.; Voelker, D.R.; Kuroki, Y. Surfactant proteins A and D bind CD14 by different mechanisms. J. Biol. Chem. 2000, 275, 22442–22451. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Head, J.; Kosma, P.; Brade, H.; Müller-Loennies, S.; Sheikh, S.; McDonald, B.; Smith, K.; Cafarella, T.; Seaton, B.; et al. Recognition of heptoses and the inner core of bacterial lipopolysaccharides by surfactant protein d. Biochemistry 2008, 47, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kuzmenko, A.; Wan, S.; Schaffer, L.; Weiss, A.; Fisher, J.H.; Kim, K.S.; McCormack, F.X. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Investig. 2003, 111, 1589–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cañadas, O.; García-Verdugo, I.; Keough, K.M.; Casals, C. SP-A permeabilizes lipopolysaccharide membranes by forming protein aggregates that extract lipids from the membrane. Biophys. J. 2008, 95, 3287–3294. [Google Scholar] [CrossRef] [PubMed]
- Keese, S.P.; Brandenburg, K.; Roessle, M.; Schromm, A.B. Pulmonary surfactant protein A-induced changes in the molecular conformation of bacterial deep-rough LPS lead to reduced activity on human macrophages. Innate Immun. 2014, 20, 787–798. [Google Scholar] [CrossRef]
- Chiba, H.; Sano, H.; Iwaki, D.; Murakami, S.; Mitsuzawa, H.; Takahashi, T.; Konishi, M.; Takahashi, H.; Kuroki, Y. Rat mannose-binding protein a binds CD14. Infect. Immun. 2001, 69, 1587–1592. [Google Scholar] [CrossRef]
- Murakami, S.; Iwaki, D.; Mitsuzawa, H.; Sano, H.; Takahashi, H.; Voelker, D.R.; Akino, T.; Kuroki, Y. Surfactant protein A inhibits peptidoglycan-induced tumor necrosis factor-alpha secretion in U937 cells and alveolar macrophages by direct interaction with Toll-like receptor 2. J. Biol. Chem. 2002, 277, 6830–6837. [Google Scholar] [CrossRef]
- Ohya, M.; Nishitani, C.; Sano, H.; Yamada, C.; Mitsuzawa, H.; Shimizu, T.; Saito, T.; Smith, K.; Crouch, E.; Kuroki, Y. Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 2006, 45, 8657–8664. [Google Scholar] [CrossRef]
- Guillot, L.; Balloy, V.; McCormack, F.X.; Golenbock, D.T.; Chignard, M.; Si-Tahar, M. Cutting edge: The immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J. Immunol. 2002, 168, 5989–5992. [Google Scholar] [CrossRef] [PubMed]
- Yamada, C.; Sano, H.; Shimizu, T.; Mitsuzawa, H.; Nishitani, C.; Himi, T.; Kuroki, Y. Surfactant Protein A Directly Interacts with TLR4 and MD-2 and Regulates Inflammatory Cellular Response. Importance of Supratrimeric Oligomerization. J. Biol. Chem. 2006, 281, 21771–21780. [Google Scholar] [CrossRef]
- Mingarro, I.; Lukovic, D.; Vilar, M.; Pérez-Gil, J. Synthetic pulmonary surfactant preparations: New developments and future trends. Curr. Med. Chem. 2008, 15, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.G.; Pérez-Gil, J. Protein-lipid interactions and surface activity in the pulmonary surfactant system. Chem. Phys. Lipids 2006, 141, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Augusto, L.A.; Li, J.; Synguelakis, M.; Johansson, J.; Chaby, R. Structural basis for interactions between lung surfactant protein C and bacterial lipopolysaccharide. J. Biol. Chem. 2002, 277, 23484–23492. [Google Scholar] [CrossRef] [PubMed]
- Augusto, L.A.; Synguelakis, M.; Espinassous, Q.; Lepoivre, M.; Johansson, J.; Chaby, R. Cellular antiendotoxin activities of lung surfactant protein C in lipid vesicles. Am. J. Respir. Crit. Care Med. 2003, 168, 335–341. [Google Scholar] [CrossRef]
- Glasser, S.W.; Senft, A.P.; Whitsett, J.A.; Maxfield, M.D.; Ross, G.F.; Richardson, T.R.; Prows, D.R.; Xu, Y.; Korfhagen, T.R. Macrophage dysfunction and susceptibility to pulmonary Pseudomonas aeruginosa infection in surfactant protein C-deficient mice. J. Immunol. 2008, 181, 621–628. [Google Scholar] [CrossRef]
- Augusto, L.A.; Synguelakis, M.; Johansson, J.; Pedron, T.; Girard, R.; Chaby, R. Interaction of pulmonary surfactant protein C with CD14 and lipopolysaccharide. Infect. Immun. 2003, 71, 61–67. [Google Scholar] [CrossRef]
- Cerutti, C.; Ridley, A.J. Endothelial Cell-Cell Adhesion and Signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelium in health and disease. Pharmacol. Rep. 2008, 60, 139–143. [Google Scholar]
- Al-Soudi, A.; Kaaij, M.H.; Tas, S.W. Endothelial Cells: From Innocent Bystanders to Active Participants in Immune Responses. Autoimmun. Rev. 2017, 16, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Janga, H.; Cassidy, L.; Wang, F.; Spengler, D.; Oestern-Fitschen, S.; Krause, M.F.; Seekamp, A.; Tholey, A.; Fuchs, S. Site-Specific and Endothelial-Mediated Dysfunction of the Alveolar-Capillary Barrier in Response to Lipopolysaccharides. J. Cell. Mol. Med. 2018, 22, 982–998. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Sanna, M. Goyert and Paul Kubes. Lipopolysaccharide-Induced Leukocyte-Endothelial Cell Interactions: A Role for CD14 Versus Toll-Like Receptor 4 Within Microvessels. J. Immunol. 2002, 169, 2111–2119. [Google Scholar] [CrossRef]
- Bannerman, D.D.; Goldblum, S.E. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L899–L914. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.H.; Chen, P.Y.; Hao, W.R.; Chiang, W.P.; Cheng, T.H.; Loh, S.H.; Leung, Y.M.; Liu, J.C.; Chen, J.J.; Sung, L.C. Lipopolysaccharide pretreatment increases protease-activated receptor-2 expression and monocyte chemoattractant protein-1 secretion in vascular endothelial cells. J. Biomed. Sci. 2017, 24, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrache, I.; Birukova, A.; Ramirez, S.I.; Garcia, J.G.; Verin, A.D. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 2003, 28, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Sawa, Y.; Ueki, T.; Hata, M.; Iwasawa, K.; Tsuruga, E.; Kojima, H.; Ishikawa, H.; Yoshida, S. LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J. Histochem. Cytochem. 2008, 56, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Bolger, M.S.; Ross, D.S.; Jiang, H.; Frank, M.M.; Ghio, A.J.; Schwartz, D.A.; Wright, J.R. Complement levels and activity in the normal and LPS-injured lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Bosmann, M.; Ward, P.A. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv. Exp. Med. Biol. 2012, 946, 147–159. [Google Scholar] [PubMed]
- Al-Sadi, R.; Ye, D.; Boivin, M.; Guo, S.; Hashimi, M.; Ereifej, L.; Ma, T.Y. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS ONE 2014, 9, e85345. [Google Scholar] [CrossRef] [PubMed]
- Reynier, F.; de Vos, A.F.; Hoogerwerf, J.J.; Bresser, P.; van der Zee, J.S.; Paye, M.; Pachot, A.; Mougin, B.; van der Poll, T. Gene Expression Profiles in Alveolar Macrophages Induced by Lipopolysaccharide in Humans. Mol. Med. 2012, 18, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Forbes, A.; Pickell, M.; Foroughian, M.; Yao, L.J.; Lewis, J.; Veldhuizen, R. Alveolar macrophage depletion is associated with increased surfactant pool sizes in adult rats. J. Appl. Physiol. 2007, 103, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussell, T.; Bell, T.J. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Jungraithmayr, W.; Kollert, F.; Goldmann, T.; Vollmer, E.; Müller-Quernheim, J.; Zissel, G. A Vicious Circle of Alveolar Macrophages and Fibroblasts Perpetuates Pulmonary Fibrosis via CCL18. Am. J. Respir. Crit. Care Med. 2006, 173, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi Monocytes Direct Alternatively Activated Profibrotic Macrophage Regulation of Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.P.; Hogaboam, C.M. Macrophages in Allergic Asthma: Fine-Tuning Their pro- and Anti-Inflammatory Actions for Disease Resolution. J. Interferon Cytokine Res. 2011, 31, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Tesfaigzi, Y. Roles of Apoptosis in Airway Epithelia. Am. J. Respir. Cell Mol. Biol 2006, 34, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Crosby, L.M.; Waters, C.M. Epithelial Repair Mechanisms in the Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Mayer, K.; Lohmeyer, J. Acute Lung Injury: How Macrophages Orchestrate Resolution of Inflammation and Tissue Repair. Front. Immunol. 2011, 2, 65. [Google Scholar] [CrossRef]
- Watford, W.T.; Wright, J.R.; Hester, C.G.; Jiang, H.; Frank, M.M. Surfactant Protein A Regulates Complement Activation. J. Immunol. 2001, 167, 6593–6600. [Google Scholar] [CrossRef] [Green Version]
- Henning, L.N.; Azad, A.K.; Parsa, K.V.; Crowther, J.E.; Tridandapani, S.; Schlesinger, L.S. Pulmonary Surfactant Protein-A regulates Toll-like receptor expression and activity in human macrophages. J. Immunol. 2008, 180, 7847–7858. [Google Scholar] [CrossRef]
- Arias-Diaz, J.; Garcia-Verdugo, I.; Casals, C.; Sanchez-Rico, N.; Vara, E.; Balibrea, J.L. Effect of surfactant protein A (SP-A) on the production of cytokines by human pulmonary macrophages. Shock 2000, 14, 300–306. [Google Scholar] [CrossRef]
- Haczku, A. Protective Role of the Lung Collectins Surfactant Protein A and Surfactant Protein D in Airway Inflammation. J. Allergy Clin. Immunol. 2008, 122, 861–879. [Google Scholar] [CrossRef]
- Bufler, P.; Schmidt, B.; Schikor, D.; Bauernfeind, A.; Crouch, E.C.; Griese, M. Surfactant protein A and D differently regulate the immune response to non-mucoid Pseudomonas aeruginosa and its lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2003, 28, 249–256. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A Critical Function for CD200 in Lung Immune Homeostasis and the Severity of Influenza Infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Adib-Conquy, M.; Moine, P.; Asehnoune, K.; Edouard, A.; Espevik, T.; Miyake, K.; Werts, C.; Cavaillon, J.M. Toll-like Receptor-mediated Tumor Necrosis Factor and Interleukin-10 Production Differ during Systemic Inflammation. Am. J. Respir. Crit. Care Med. 2003, 168, 158–164. [Google Scholar] [CrossRef]
- Chen, L.C.; Gordon, R.E.; Laskin, J.D.; Laskin, D.L. Role of TLR-4 in Liver Macrophage and Endothelial Cell Responsiveness During Acute Endotoxemia. Exp. Mol. Pathol. 2007, 83, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Hoogerwerf, J.J.; de Vos, A.F.; Bresser, P.; van der Zee, J.S.; Pater, J.M.; de Boer, A.; Tanck, M.; Lundell, D.L.; Her-Jenh, C.; Draing, C.; et al. Lung Inflammation Induced by Lipoteichoic Acid or Lipopolysaccharide in Humans. Am. J. Respir. Crit. Care Med. 2008, 178, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Pröll, M.; Hölker, M.; Tholen, E.; Tesfaye, D.; Looft, C.; Schellander, K.; Cinar, M.U. Alveolar Macrophage Phagocytic Activity Is Enhanced with LPS Priming, and Combined Stimulation of LPS and Lipoteichoic Acid Synergistically Induce pro-Inflammatory Cytokines in Pigs. Innate Immun. 2013, 19, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Cinar, M.U.; Uddin, M.J.; Tholen, E.; Tesfaye, D.; Looft, C.; Schellander, K. Expression of Toll-like receptors and downstream genes in lipopolysaccharide-induced porcine alveolar macrophages. Vet. Immunol. Immunopathol. 2012, 146, 62–73. [Google Scholar] [CrossRef]
- Feng, X.; Deng, T.; Zhang, Y.; Su, S.; Wei, C.; Han, D. Lipopolysaccharide inhibits macrophage phagocytosis of apoptotic neutrophils by regulating the production of tumour necrosis factor alpha and growth arrest-specific gene 6. Immunology 2011, 132, 287–295. [Google Scholar] [CrossRef]
- Vaccaro, C.; Brody, J.S. Ultrastructure of developing alveoli. I. The role of the interstitial fibroblast. Anat. Rec. 1978, 192, 467–479. [Google Scholar] [CrossRef]
- Ruiz-Camp, J.; Morty, R.E. Divergent fibroblast growth factor signaling pathways in lung fibroblast subsets: Where do we go from here? Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, 751–755. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: one function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhu, Y.; Jiang, H. Toll-like receptor 4 mediates lipopolysaccharide-induced collagen secretion by phosphoinositide3-kinase-Akt pathway in fibroblasts during acute lung injury. J. Recept. Signal Transduct. Res. 2009, 29, 119–125. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gao, Y.; Deng, Y.; Li, W.; Chen, Y.; Xing, S.; Zhao, X.; Ding, J.; Wang, X. Lipopolysaccharide induces lung fibroblast proliferation through Toll-like receptor 4 signaling and the phosphoinositide3-kinase-Akt pathway. PLoS ONE 2012, 7, e35926. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, R.; Hu, J.; Mo, Z.; Yang, Z.; Liao, D.; Zhong, N. Attenuation of bleomycin-induced lung fibrosis by oxymatrine is associated with regulation of fibroblast proliferation and collagen production in primary culture. Basic Clin. Pharmacol. Toxicol. 2008, 103, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Diebold, D.; Nho, R.; Perlman, D.; Kleidon, J.; Kahm, J.; Avdulov, S.; Peterson, M.; Nerva, J.; Bitterman, P.; et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J. Exp. Med. 2008, 205, 1659–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Zhu, Y.; Jiang, H. Inhibiting toll-like receptor 4 signaling ameliorates pulmonary fibrosis during acute lung injury induced by lipopolysaccharide: An experimental study. Respir. Res. 2009, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nova, Z.; Skovierova, H.; Calkovska, A. Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury. Int. J. Mol. Sci. 2019, 20, 831. https://doi.org/10.3390/ijms20040831
Nova Z, Skovierova H, Calkovska A. Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury. International Journal of Molecular Sciences. 2019; 20(4):831. https://doi.org/10.3390/ijms20040831
Chicago/Turabian StyleNova, Zuzana, Henrieta Skovierova, and Andrea Calkovska. 2019. "Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury" International Journal of Molecular Sciences 20, no. 4: 831. https://doi.org/10.3390/ijms20040831
APA StyleNova, Z., Skovierova, H., & Calkovska, A. (2019). Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury. International Journal of Molecular Sciences, 20(4), 831. https://doi.org/10.3390/ijms20040831