Cholesterol and the Safety Factor for Neuromuscular Transmission


Abstract
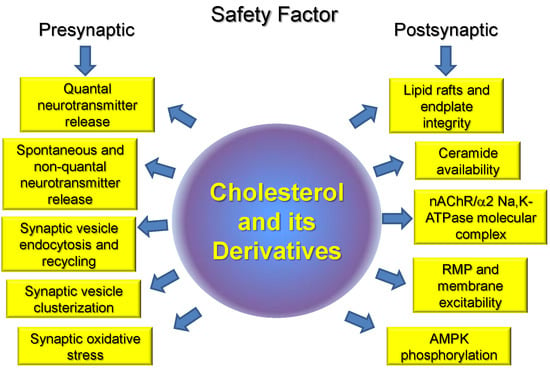
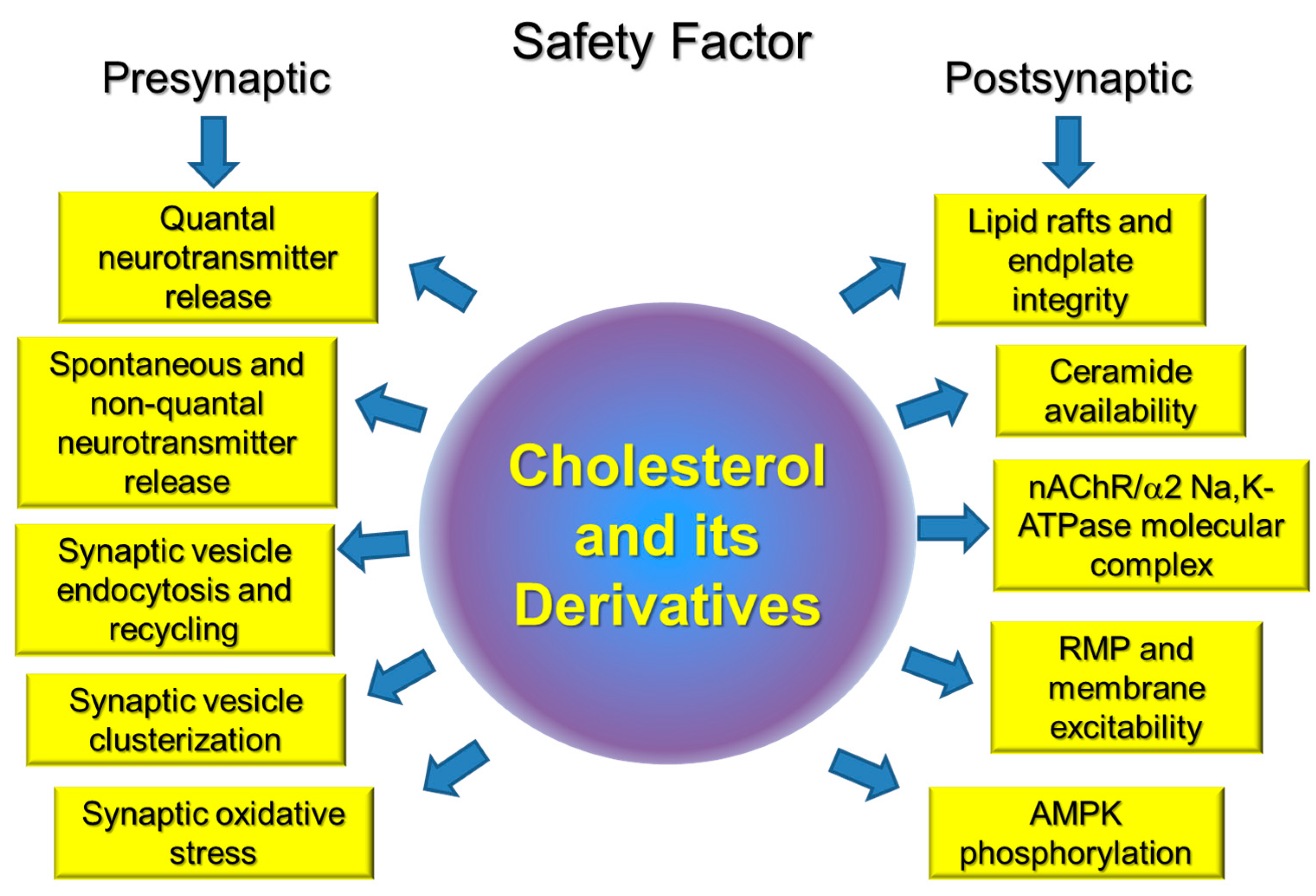
:
1. Introduction
2. Cholesterol Production
3. Cholesterol and Quantal Neurotransmission Release
4. Effects of Cholesterol Derivatives on Neurotransmitter Release and Synaptic Vesicle Cycle
5. Cholesterol and Proteins Involved in Synaptic Vesicle Cycle
6. Cholesterol–Na,K-ATPase Interactions
7. Cholesterol–nAChR Interactions
8. Cholesterol and Motor Dysfunction
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-BTX | rhodamine-conjugated α-bungarotoxin |
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| ALS | amyotrophic lateral sclerosis |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| AP | action potential |
| ATP | adenosine triphosphate |
| AZs | active zones |
| 5Ch3 | 5α-cholestan-3-one |
| CSP | cysteine string protein |
| CTxB | cholera toxin B subunit |
| EPP | endplate potential |
| GPCRs | G-protein coupled receptors |
| 24HC | 24S-hydroxycholesterol |
| HS | hindlimb suspension |
| LXR | liver X receptor |
| MβCD | methyl-β-cyclodextrin |
| MEPP | miniature endplate potential |
| nAChR | nicotinic acetylcholine receptor |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NMJ | neuromuscular junction |
| RMP | resting membrane potential |
| ROS | reactive oxygen species |
| SOD1G93A | superoxide dismutase 1 |
| SV | synaptic vesicle |
| VGLUT | vesicular glutamate transporter |
References
- Wood, S.J.; Slater, C.R. Safety factor at the neuromuscular junction. Prog. Neurobiol. 2001, 64, 393–429. [Google Scholar] [CrossRef]
- Ruff, R.L. Endplate contributions to the safety factor for neuromuscular transmission. Muscle Nerve 2011, 44, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, F.; Waites, C.L.; Garner, C.C. Presynaptic active zones in invertebrates and vertebrates. EMBO Rep. 2015, 16, 923–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanes, J.S.; Lichtman, J.W. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999, 22, 389–442. [Google Scholar] [CrossRef] [PubMed]
- Filatov, G.N.; Pinter, M.J.; Rich, M.M. Resting Potential-dependent Regulation of the Voltage Sensitivity of Sodium Channel Gating in Rat Skeletal Muscle In Vivo. J. Gen. Physiol. 2005, 126, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.T.; Cottey, E.; Cottey, A.; Stefanski, C.; Carlson, C.G. Reduced resting potentials in dystrophic (mdx) muscle fibers are secondary to NF-κB-dependent negative modulation of ouabain sensitive Na+-K+ pump activity. J. Neurosci. 2011, 303, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Ruff, R.L.; Leigh, R.J. Neuromuscular transmission failure in myasthenia gravis: Decrement of safety factor and susceptibility of extraocular muscles. Ann. N. Y. Acad. Sci. 2012, 1275, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiøtt, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim. Biophys. Acta 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belani, J.D. Chirality Effect on Cholesterol Modulation of Protein Function. Adv. Exp. Med. Biol. 2019, 1115, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Mohler, E.R., 3rd; Tian, A.; Baumgart, T.; Diamond, S.L. Membrane cholesterol is a biomechanical regulator of neutrophil adhesion. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Oh, H.G.; Park, M.K.; Kim, T.W.; Chung, S. Increasing Membrane Cholesterol Level Increases the Amyloidogenic Peptide by Enhancing the Expression of Phospholipase C. J. Neurodegener. Dis. 2013, 2013, 407903. [Google Scholar] [CrossRef] [PubMed]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef] [PubMed]
- Amsalem, M.; Poilbout, C.; Ferracci, G.; Delmas, P.; Padilla, F. Membrane cholesterol depletion as a trigger of Nav1.9 channel-mediated inflammatory pain. EMBO J. 2018, 37, e97349. [Google Scholar] [CrossRef] [PubMed]
- Meleleo, D.; Sblano, C. Influence of cholesterol on human calcitonin channel formation. Possible role of sterol as molecular chaperone. AIMS Biophys. 2019, 6, 23–38. [Google Scholar] [CrossRef]
- Arenas, F.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Stumpf, S.K. Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 2015, 1851, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Funfschilling, U.; Jockusch, W.J.; Sivakumar, N.; Mobius, W.; Corthals, K.; Li, S.; Quintes, S.; Kim, Y.; Schaap, I.A.; Rhee, J.S.; et al. Critical time window of neuronal cholesterol synthesis during neurite outgrowth. J. Neurosci. 2012, 32, 7632–7645. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Kasimov, M.R.; Zefirov, A.L. Brain Cholesterol Metabolism and Its Defects: Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. Acta Nat. 2016, 8, 58–73. [Google Scholar]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef] [PubMed]
- Comley, L.H.; Fuller, H.R.; Wishart, T.M.; Mutsaers, C.A.; Thomson, D.; Wright, A.K.; Ribchester, R.R.; Morris, G.E.; Parson, S.H.; Horsburgh, K.; et al. ApoE isoform-specific regulation of regeneration in the peripheral nervous system. Hum. Mol. Genet. 2011, 20, 2406–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.Y.; Liu, Y.; Tennert, C.; Sugiura, Y.; Karakatsani, A.; Kröger, S.; Johnson, E.B.; Hammer, R.E.; Lin, W.; Herz, J. APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. eLife 2013, 2, e00220. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Seo, T.; Park, T.; Yagyu, H.; Hu, Y.; Son, N.H.; Augustus, A.S.; Vikramadithyan, R.K.; Ramakrishnan, R.; Pulawa, L.K.; et al. Effects of lipoprotein lipase and statins on cholesterol uptake into heart and skeletal muscle. J. Lipid Res. 2007, 48, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, G.; Sánchez-Aguilera, P.; Jaimovich, E.; Hidalgo, C.; Llanos, P. Membrane Cholesterol in Skeletal Muscle: A Novel Player in Excitation-Contraction Coupling and Insulin Resistance. J. Diabetes Res. 2017, 2017, 3941898. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Tibolla, G.; Catapano, A.L. Statins and skeletal muscles toxicity: From clinical trials to everyday practice. Pharmacol. Res. 2014, 88, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, R.C.; Johnson, K.M. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J. Biol. Chem. 2008, 283, 15521–15525. [Google Scholar] [CrossRef] [PubMed]
- Archer, A.; Laurencikiene, J.; Ahmed, O.; Steffensen, K.R.; Parini, P.; Gustafsson, J.A.; Korach-André, M. Skeletal muscle as a target of LXR agonist after long-term treatment: Focus on lipid homeostasis. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E494–E502. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.; Hughes, M.G.; Thomas, A.W.; Morris, K. The Ability of Exercise-Associated Oxidative Stress to Trigger Redox-Sensitive Signalling Responses. Antioxidants 2017, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O. Synaptic vesicle recycling: Steps and principles. EMBO J. 2014, 33, 788–822. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, S.O.; Betz, W.J. Synaptic vesicle pools. Nat. Rev. Neurosci. 2005, 6, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, Y.; Bridgman, P.C. Absence of filipin-sterol complexes from the membranes of active zones and acetylcholine receptor aggregates at frog neuromuscular junctions. J. Cell. Biol. 1981, 88, 453–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.P.; Propst, J.W. Absence of sterol-specific complexes at active zones of degenerating and regenerating frog neuromuscular junctions. J. Neurocytol. 1986, 15, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Zamir, O.; Charlton, M.P. Cholesterol and synaptic transmitter release at crayfish neuromuscular junctions. J. Physiol. 2006, 571, 83–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrov, A.M.; Kudryashova, K.E.; Odnoshivkina, Y.G.; Zefirov, A.L. Cholesterol and lipid rafts in the plasma membrane of nerve terminal and membrane of synaptic vesicles. Neurochem. J. 2011, 5, 13–19. [Google Scholar] [CrossRef]
- Petrov, A.M.; Naumenko, N.V.; Uzinskaya, K.V.; Giniatullin, A.R.; Urazaev, A.K.; Zefirov, A.L. Increased non-quantal release of acetylcholine after inhibition of endocytosis by methyl-β-cyclodextrin: The role of vesicular acetylcholine transporter. Neuroscience 2011, 186, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kasimov, M.R.; Giniatullin, A.R.; Zefirov, A.L.; Petrov, A.M. Effects of 5α-cholestan-3-one on the synaptic vesicle cycle at the mouse neuromuscular junction. Biochim. Biophys. Acta 2015, 1851, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Kasimov, M.R.; Zakyrjanova, G.F.; Giniatullin, A.R.; Zefirov, A.L.; Petrov, A.M. Similar oxysterols may lead to opposite effects on synaptic transmission: Olesoxime versus 5 α-cholestan-3-one at the frog neuromuscular junction. Biochim. Biophys. Acta 2016, 1861, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Mukhutdinova, K.A.; Kasimov, M.R.; Giniatullin, A.R.; Zakyrjanova, G.F.; Petrov, A.M. 24S-hydroxycholesterol suppresses neuromuscular transmission in SOD1(G93A) mice: A possible role of NO and lipid rafts. Mol. Cell. Neurosci. 2018, 88, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Kasimov, M.R.; Giniatullin, A.R.; Tarakanova, O.I.; Zefirov, A.L. The role of cholesterol in the exo- and endocytosis of synaptic vesicles in frog motor nerve endings. Neurosci. Behav. Physiol. 2010, 40, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Yakovleva, A.A.; Zefirov, A.L. Role of membrane cholesterol in spontaneous exocytosis at frog neuromuscular synapses: Reactive oxygen species-calcium interplay. J. Physiol. 2014, 592, 4995–5009. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Zakyrjanova, G.F.; Yakovleva, A.A.; Zefirov, A.L. Inhibition of protein kinase C affects on mode of synaptic vesicle exocytosis due to cholesterol depletion. Biochem. Biophys. Res. Commun. 2015, 456, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Tarakanova, O.I.; Petrov, A.M.; Zefirov, A.L. The role of membrane cholesterol in neurotransmitter release from motor nerve terminals. Dokl. Biol. Sci. 2011, 438, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, H.A.; Lima, R.F.; Fonseca Mde, C.; Amaral, E.A.; Martinelli, P.M.; Naves, L.A.; Gomez, M.V.; Kushmerick, C.; Prado, M.A.; Guatimosim, C. Membrane cholesterol regulates different modes of synaptic vesicle release and retrieval at the frog neuromuscular junction. Eur. J. Neurosci. 2013, 38, 2978–2987. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Vieira, L.B.; Gomez, M.V.; Guatimosim, C. Cholesterol as a key player in the balance of evoked and spontaneous glutamate release in rat brain cortical synaptosomes. Neurochem. Int. 2012, 61, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyagarajan, B.; Potian, J.G.; McArdle, J.J.; Baskaran, P. Perturbation to Cholesterol at the Neuromuscular Junction Confers Botulinum Neurotoxin A Sensitivity to Neonatal Mice. Toxicol. Sci. 2017, 159, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Sugita, S.; Charlton, M.P. Cholesterol-dependent kinase activity regulates transmitter release from cerebellar synapses. J. Neurosci. 2010, 30, 6116–6121. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Dason, J.S.; Smith, A.J.; Marin, L.; Charlton, M.P. Vesicular sterols are essential for synaptic vesicle cycling. J. Neurosci. 2010, 30, 15856–15865. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.Y.; Xu, J. Cholesterol regulates multiple forms of vesicle endocytosis at a mammalian central synapse. J. Neurochem. 2015, 134, 247–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dason, J.S.; Smith, A.J.; Marin, L.; Charlton, M.P. Cholesterol and F-actin are required for clustering of recycling synaptic vesicle proteins in the presynaptic plasma membrane. J. Physiol. 2014, 592, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, C.; Hannah, M.J.; Fahrenholz, F.; Huttner, W.B. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell. Biol. 2000, 2, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Fewou, S.N.; Rupp, A.; Nickolay, L.E.; Carrick, K.; Greenshields, K.N.; Pediani, J.; Plomp, J.J.; Willison, H.J. Anti-ganglioside antibody internalization attenuates motor nerve terminal injury in a mouse model of acute motor axonal neuropathy. J. Clin. Investig. 2012, 122, 1037–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrov, A.M.; Kasimov, M.R.; Zefirov, A.L. Cholesterol in the Pathogenesis of Alzheimer’s, Parkinson’s Diseases and Autism: Link to Synaptic Dysfunction. Acta Nat. 2017, 9, 26–37. [Google Scholar]
- Testa, G.; Rossin, D.; Poli, G.; Biasi, F.; Leonarduzzi, G. Implication of oxysterols in chronic inflammatory human diseases. Biochimie 2018, 153, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Kasimov, M.R.; Giniatullin, A.R.; Zefirov, A.L. Effects of Oxidation of Membrane Cholesterol on the Vesicle Cycle in Motor Nerve Terminals in the Frog Rana Ridibunda. Neurosci. Behav. Physiol. 2014, 44, 1020–1030. [Google Scholar] [CrossRef]
- Kasimov, M.R.; Fatkhrakhmanova, M.R.; Mukhutdinova, K.A.; Petrov, A.M. 24S-Hydroxycholesterol enhances synaptic vesicle cycling in the mouse neuromuscular junction: Implication of glutamate NMDA receptors and nitric oxide. Neuropharmacology 2017, 117, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.M.; Mallozzi, C.; Macchia, G.; Maura, G.; Petrucci, T.C.; Minetti, M. Peroxynitrite affects exocytosis and SNARE complex formation and induces tyrosine nitration of synaptic proteins. J. Neurochem. 2002, 82, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Robitaille, R. Differential frequency-dependent regulation of transmitter release by endogenous nitric oxide at the amphibian neuromuscular synapse. J. Neurosci. 2001, 21, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.W.; Bourgognon, J.M.; Spiers, J.G.; Breda, C.; Campesan, S.; Butcher, A.; Mallucci, G.R.; Dinsdale, D.; Morone, N.; Mistry, R.; et al. Nitric oxide-mediated posttranslational modifications control neurotransmitter release by modulating complexin farnesylation and enhancing its clamping ability. PLoS Biol. 2018, 16, e2003611. [Google Scholar] [CrossRef] [PubMed]
- DeBarber, A.E.; Sandlers, Y.; Pappu, A.S.; Merkens, L.S.; Duell, P.B.; Lear, S.R.; Erickson, S.K.; Steiner, R.D. Profiling sterols in cerebrotendinous xanthomatosis: Utility of Girard derivatization and high resolution exact mass LC-ESI-MS(n) analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. IDrugs 2010, 13, 568–580. [Google Scholar] [PubMed]
- Gouarné, C.; Tracz, J.; Paoli, M.G.; Deluca, V.; Seimandi, M.; Tardif, G.; Xilouri, M.; Stefanis, L.; Bordet, T.; Pruss, R.M. Protective role of olesoxime against wild-type α-synuclein-induced toxicity in human neuronally differentiated SHSY-5Y cells. Br. J. Pharmacol. 2015, 172, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Magalon, K.; Le Grand, M.; El Waly, B.; Moulis, M.; Pruss, R.; Bordet, T.; Cayre, M.; Belenguer, P.; Carré, M.; Durbec, P. Olesoxime favors oligodendrocyte differentiation through a functional interplay between mitochondria and microtubules. Neuropharmacology 2016, 111, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.J.; Ortiz Rios, M.M.; Riess, O.; Clemens, L.E.; Nguyen, H.P. The calpain-suppressing effects of olesoxime in Huntington’s disease. Rare Dis. 2016, 4, e1153778. [Google Scholar] [CrossRef] [PubMed]
- Taverna, E.; Saba, E.; Rowe, J.; Francolini, M.; Clementi, F.; Rosa, P. Role of lipid microdomains in P/Q-type calcium channel (Cav2.1) clustering and function in presynaptic membranes. J. Biol. Chem. 2004, 279, 5127–5134. [Google Scholar] [CrossRef] [PubMed]
- Thoreson, W.B.; Mercer, A.J.; Cork, K.M.; Szalewski, R.J. Lateral mobility of L-type calcium channels in synaptic terminals of retinal bipolar cells. Mol. Vis. 2013, 19, 16–24. [Google Scholar] [PubMed]
- Ronzitti, G.; Bucci, G.; Emanuele, M.; Leo, D.; Sotnikova, T.D.; Mus, L.V.; Soubrane, C.H.; Dallas, M.L.; Thalhammer, A.; Cingolani, L.A.; et al. Exogenous α-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 2014, 34, 10603–10615. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.H.; He, L.; Sui, S.F. Lipid rafts association of synaptotagmin I on synaptic vesicles. Biochemistry 2008, 73, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Matthies, H.J.; Han, Q.; Shields, A.; Wright, J.; Moore, J.L.; Winder, D.G.; Galli, A.; Blakely, R.D. Subcellular localization of the antidepressant-sensitive norepinephrine transporter. BMC Neurosci. 2009, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- De Juan-Sanz, J.; Núñez, E.; Zafra, F.; Berrocal, M.; Corbacho, I.; Ibáñez, I.; Arribas-González, E.; Marcos, D.; López-Corcuera, B.; Mata, A.M.; et al. Presynaptic control of glycine transporter 2 (GlyT2) by physical and functional association with plasma membrane Ca2+-ATPase (PMCA) and Na+-Ca2+ exchanger (NCX). J. Biol. Chem. 2014, 289, 34308–34324. [Google Scholar] [CrossRef] [PubMed]
- Rahbek-Clemmensen, T.; Lycas, M.D.; Erlendsson, S.; Eriksen, J.; Apuschkin, M.; Vilhardt, F.; Jørgensen, T.N.; Hansen, F.H.; Gether, U. Super-resolution microscopy reveals functional organization of dopamine transporters into cholesterol and neuronal activity-dependent nanodomains. Nat. Commun. 2017, 8, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinaka, K.; Kumanogoh, H.; Nakamura, S.; Maekawa, S. Identification of V-ATPase as a major component in the raft fraction prepared from the synaptic plasma membrane and the synaptic vesicle of rat brain. Neurosci. Lett. 2004, 363, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Cho, W.J.; Shin, L.; Jena, B.P. Involvement of cholesterol in synaptic vesicle swelling. Exp. Biol. Med. 2010, 235, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.H.; Tamm, L.K. Molecular mechanism of cholesterol- and polyphosphoinositide-mediated syntaxin clustering. Biochemistry 2011, 50, 9014–9022. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Cubí, R.; Blasi, J.; Aguilera, J. Synaptic proteins associate with a sub-set of lipid rafts when isolated from nerve endings at physiological temperature. Biochem. Biophys. Res. Commun. 2006, 348, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.J.; Jeremic, A.; Jin, H.; Ren, G.; Jena, B.P. Neuronal fusion pore assembly requires membrane cholesterol. Cell Biol. Int. 2007, 31, 1301–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreutzberger, A.J.; Kiessling, V.; Tamm, L.K. High cholesterol obviates a prolonged hemifusion intermediate in fast SNARE-mediated membrane fusion. Biophys. J. 2015, 109, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Kyung, J.W.; Kim, J.M.; Lee, W.; Ha, T.Y.; Cha, S.H.; Chung, K.H.; Choi, D.J.; Jou, I.; Song, W.K.; Joe, E.H.; et al. DJ-1 deficiency impairs synaptic vesicle endocytosis and reavailability at nerve terminals. Proc. Natl. Acad. Sci. USA 2018, 115, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.T.; Ryoo, K.; Lin, A.; Janoschka, S.R.; Augustine, G.J.; Porton, B. Synapsins regulate brain-derived neurotrophic factor-mediated synaptic potentiation and axon elongation by acting on membrane rafts. Eur. J. Neurosci. 2017, 45, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Moral-Naranjo, M.T.; Montenegro, M.F.; Muñoz-Delgado, E.; Campoy, F.J.; Vidal, C.J. The levels of both lipid rafts and raft-located acetylcholinesterase dimers increase in muscle of mice with muscular dystrophy by merosin deficiency. Biochim. Biophys. Acta 2010, 1802, 754–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenegro, M.F.; Cabezas-Herrera, J.; Campoy, F.J.; Muñoz-Delgado, E.; Vidal, C.J. Lipid rafts of mouse liver contain nonextended and extended acetylcholinesterase variants along with M3 muscarinic receptors. FASEB J. 2017, 31, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Giniatullin, A.; Petrov, A.; Giniatullin, R. The involvement of P2Y12 receptors, NADPH oxidase, and lipid rafts in the action of extracellular ATP on synaptic transmission at the frog neuromuscular junction. Neuroscience 2015, 285, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, S.; Di Scala, C.; Wang, Q.; Wandall, H.H.; Fantini, J.; Zhang, Y.Q. The glycosphingolipid MacCer promotes synaptic bouton formation in Drosophila by interacting with Wnt. eLife 2018, 7, e38183. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T. Na+-K+ pump regulation and skeletal muscle contractility. Physiol. Rev. 2003, 83, 1269–1324. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: Functional significance. J. Gen. Physiol. 2013, 142, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F655. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Avila, J.; Cózar-Castellano, I.; Brownleader, M.D.; Trevan, M.; Francis, M.J.; Lamb, J.F.; Martín-Vasallo, P. Na+,K+-ATPase isozyme diversity; comparative biochemistry and physiological implications of novel functional interactions. Biosci. Rep. 2000, 20, 51–91. [Google Scholar] [CrossRef] [PubMed]
- Sejersted, O.M.; Sjogaard, G. Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol. Rev. 2000, 80, 1411–1481. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Singh, D.K.; Rosenhouse-Dantsker, A. Cholesterol binding to ion channels. Front. Physiol. 2014, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, F.; Habeck, M.; Kanai, R.; Toyoshima, C.; Karlish, S.J. General and specific lipid-protein interactions in Na,K-ATPase. Biochim. Biophys. Acta 2015, 1848, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Poguzhelskaya, E.E.; Antonov, S.M. Downregulation of calcium-dependent NMDA receptor desensitization by sodium-calcium exchangers: A role of membrane cholesterol. BMC Neurosci. 2018, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Haviv, H.; Habeck, M.; Kanai, R.; Toyoshima, C.; Karlish, S.J. Neutral phospholipids stimulate Na,K-ATPase activity: A specific lipid-protein interaction. J. Biol. Chem. 2013, 288, 10073–10081. [Google Scholar] [CrossRef] [PubMed]
- Matchkov, V.V.; Krivoi, I.I. Specialized functional diversity and interactions of the Na,K-ATPase. Front. Physiol. 2016, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlowski, J.; Lingrel, J.B. Tissue-specific and developmental regulation of rat Na,K-ATPase catalytic α isoform and β subunit mRNAs. J. Biol. Chem. 1988, 263, 10436–10442. [Google Scholar] [PubMed]
- He, S.; Shelly, D.A.; Moseley, A.E.; James, P.F.; James, J.H.; Paul, R.J.; Lingrel, J.B. The α1- and α2-isoforms of Na-K-ATPase play different roles in skeletal muscle contractility. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R917–R925. [Google Scholar] [CrossRef] [PubMed]
- Heiny, J.A.; Kravtsova, V.V.; Mandel, F.; Radzyukevich, T.L.; Benziane, B.; Prokofiev, A.V.; Pedersen, S.E.; Chibalin, A.V.; Krivoi, I.I. The nicotinic acetylcholine receptor and the Na,K-ATPase α2 isoform interact to regulate membrane electrogenesis in skeletal muscle. J. Biol. Chem. 2010, 285, 28614–28626. [Google Scholar] [CrossRef] [PubMed]
- Radzyukevich, T.L.; Neumann, J.C.; Rindler, T.N.; Oshiro, N.; Goldhamer, D.J.; Lingrel, J.B.; Heiny, J.A. Tissue-specific role of the Na,K-ATPase α2 isozyme in skeletal muscle. J. Biol. Chem. 2013, 288, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- DiFranco, M.; Hakimjavadi, H.; Lingrel, J.B.; Heiny, J.A. Na,K-ATPase α2 activity in mammalian skeletal muscle T-tubules is acutely stimulated by extracellular K+. J. Gen. Physiol. 2015, 146, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova, V.V.; Petrov, A.M.; Matchkov, V.V.; Bouzinova, E.V.; Vasiliev, A.N.; Benziane, B.; Zefirov, A.L.; Chibalin, A.V.; Heiny, J.A.; Krivoi, I.I. Distinct α2 Na,K-ATPase membrane pools are differently involved in early skeletal muscle remodeling during disuse. J. Gen. Physiol. 2016, 147, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Kutz, L.C.; Mukherji, S.T.; Wang, X.; Bryant, A.; Larre, I.; Heiny, J.A.; Lingrel, J.B.; Pierre, S.V.; Xie, Z. Isoform-specific role of Na/K-ATPase α1 in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E620–E629. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xiong, W.C.; Mei, L. Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J. Neurosci. 2006, 26, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saks, V.; Monge, C.; Guzun, R. Philosophical basis and some historical aspects of systems biology: From Hegel to Noble—Applications for bioenergetic research. Int. J. Mol. Sci. 2009, 10, 1161–1192. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Xie, Z. The Na-K-ATPase and calcium-signaling microdomains. Physiology 2008, 23, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Morrill, G.A.; Kostellow, A.B.; Askari, A. Caveolin-Na/K-ATPase interactions: Role of transmembrane topology in non-genomic steroid signal transduction. Steroids 2012, 77, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22, 990. [Google Scholar] [CrossRef]
- Wang, H.; Haas, M.; Liang, M.; Cai, T.; Tian, J.; Li, S.; Xie, Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J. Biol. Chem. 2004, 279, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Wang, H.; Chen, Y.; Liu, L.; Gunning, W.T.; Quintas, L.E.; Xie, Z.J. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J. Cell Biol. 2008, 182, 1153–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From Cell Biology to Animal Physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, T.; Wang, H.; Li, Z.; Loreaux, E.; Lingrel, J.B.; Xie, Z. Regulation of intracellular cholesterol distribution by Na/K-ATPase. J. Biol. Chem. 2009, 284, 14881–14890. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Ye, Q.; Tian, J.; Jing, R.; Xie, Z. Regulation of α1 Na/K-ATPase expression by cholesterol. J. Biol. Chem. 2011, 286, 15517–15524. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova, V.V.; Petrov, A.M.; Vasiliev, A.N.; Zefirov, A.L.; Krivoi, I.I. Role of cholesterol in the maintenance of endplate electrogenesis in rat diaphragm. Bull. Exp. Biol. Med. 2015, 158, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.M.; Kravtsova, V.V.; Matchkov, V.V.; Vasiliev, A.N.; Zefirov, A.L.; Chibalin, A.V.; Heiny, J.A.; Krivoi, I.I. Membrane lipid rafts are disturbed in the response of rat skeletal muscle to short-term disuse. Am. J. Physiol. Cell Physiol. 2017, 312, C627–C637. [Google Scholar] [CrossRef] [PubMed]
- Habeck, M.; Haviv, H.; Katz, A.; Kapri-Pardes, E.; Ayciriex, S.; Shevchenko, A.; Ogawa, H.; Toyoshima, C.; Karlish, S.J. Stimulation, inhibition, or stabilization of Na,K-ATPase caused by specific lipid interactions at distinct sites. J. Biol. Chem. 2015, 290, 4829–4842. [Google Scholar] [CrossRef] [PubMed]
- Lifshitz, Y.; Petrovich, E.; Haviv, H.; Goldshleger, R.; Tal, D.M.; Garty, H.; Karlish, S.J.D. Purification of the human α2 isoform of Na,K-ATPase expressed in Pichia pastoris. Stabilization by lipids and FXYD1. Biochemistry 2007, 46, 14937–14950. [Google Scholar] [CrossRef] [PubMed]
- Kapri-Pardes, E.; Katz, A.; Haviv, H.; Mahmmoud, Y.; Ilan, M.; Khalfin-Penigel, I.; Carmeli, S.; Yarden, O.; Karlish, S.J.D. Stabilization of the α2 isoform of Na,K-ATPase by mutations in a phospholipid binding pocket. J. Biol. Chem. 2011, 286, 42888–42899. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Sooksawate, T.; Simmonds, M.A. Effects of membrane cholesterol on the sensitivity of the GABAA receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology 2001, 40, 178–184. [Google Scholar] [CrossRef]
- Eroglu, C.; Brugger, B.; Wieland, F.; Sinning, I. Glutamate-binding affinity of Drosophila metabotropic glutamate receptor is modulated by association with lipid rafts. Proc. Natl. Acad. Sci. USA 2003, 100, 10219–10224. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, J.E.; Domville, J.A.; Therien, J.P.D. The Role of Cholesterol in the Activation of Nicotinic Acetylcholine Receptors. Curr. Top. Membr. 2017, 80, 95–137. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Gimpl, G.; Fahrenholz, F. Regulation of receptor function by cholesterol. Cell. Mol. Life Sci. 2000, 57, 1577–1592. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, G.; Hénin, J.; Law, R.; Eckenhoff, R.; Klein, M.L. Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 14418–14423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrantes, F.J. Cholesterol and nicotinic acetylcholine receptor: An intimate nanometer-scale spatial relationship spanning the billion year time-scale. Biomed. Spectrosc. Imaging 2016, 5, S67–S86. [Google Scholar] [CrossRef]
- Scher, M.G.; Bloch, R.J. The lipid bilayer of acetylcholine receptor clusters of cultured rat myotubes is organized into morphologically distinct domains. Exp. Cell. Res. 1991, 195, 79–91. [Google Scholar] [CrossRef]
- Marchand, S.; Devillers-Thiéry, A.; Pons, S.; Changeux, J.P.; Cartaud, J. Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts. J. Neurosci. 2002, 22, 8891–8901. [Google Scholar] [CrossRef] [PubMed]
- Campagna, J.A.; Fallon, J. Lipid rafts are involved in C95 (4,8) agrin fragment-induced acetylcholine receptor clustering. Neuroscience 2006, 138, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Stetzkowski-Marden, F.; Gaus, K.; Recouvreur, M.; Cartaud, A.; Cartaud, J. Agrin elicits membrane lipid condensation at sites of acetylcholine receptor clusters in C2C12 myotubes. J. Lipid Res. 2006, 47, 2121–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Moreno, J.; Tirado-Cortes, A.; Álvarez, R.; Irles, C.; Mas-Oliva, J.; Ortega, A. Cholesterol depletion uncouples β-dystroglycans from discrete sarcolemmal domains, reducing the mechanical activity of skeletal muscle. Cell Physiol. Biochem. 2012, 29, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Pato, C.; Stetzkowski-Marden, F.; Gaus, K.; Recouvreur, M.; Cartaud, A.; Cartaud, J. Role of lipid rafts in agrin-elicited acetylcholine receptor clustering. Chem. Biol. Interact. 2008, 175, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Nikolsky, E.E.; Zemkova, H.; Voronin, V.A.; Vyskocil, F. Role of non-quantal acetylcholine release in surplus polarization of mouse diaphragm fibres at the endplate zone. J. Physiol. 1994, 477, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Vyskocil, F.; Malomouzh, A.I.; Nikolsky, E.E. Non-quantal acetylcholine release at the neuromuscular junction. Physiol. Res. 2009, 58, 763–784. [Google Scholar] [PubMed]
- Krivoi, I.I.; Drabkina, T.M.; Kravtsova, V.V.; Vasiliev, A.N.; Eaton, M.J.; Skatchkov, S.N.; Mandel, F. On the functional interaction between nicotinic acetylcholine receptor and Na+,K+-ATPase. Pflugers Arch. 2006, 452, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Sun, H.; Xiao, Y.; Zhang, Y.; Wang, X.; Xu, X.; Liu, Z.; Fang, J.; Li, Z. Functional interaction of nicotinic acetylcholine receptors and Na+/K+ ATPase from Locusta migratoria manilensis (Meyen). Sci. Rep. 2015, 5, 8849. [Google Scholar] [CrossRef] [PubMed]
- Hezel, M.; de Groat, W.C.; Galbiati, F. Caveolin-3 promotes nicotinic acetylcholine receptor clustering and regulates neuromuscular junction activity. Mol. Biol. Cell 2010, 21, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Razani, B.; Lisanti, M.P. Caveolae and caveolin-3 in muscular dystrophy. Trends Mol. Med. 2001, 7, 435–441. [Google Scholar] [CrossRef]
- Prince, R.J.; Sine, S.M. Acetylcholine and epibatidine binding to muscle acetylcholine receptors distinguish between concerted and uncoupled models. J. Biol. Chem. 1999, 274, 19623–19629. [Google Scholar] [CrossRef] [PubMed]
- Mourot, A.; Rodrigo, J.; Kotzyba-Hibert, F.; Bertrand, S.; Bertrand, D.; Goeldner, M. Probing the reorganization of the nicotinic acetylcholine receptor during desensitization by time-resolved covalent labeling using [3H]AC5, a photoactivatable agonist. Mol. Pharmacol. 2006, 69, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Suzuki, K.; Higuchi, M.; Balogh, L.; Boldogh, I.; Koltai, E. Physical exercise, reactive oxygen species and neuroprotection. Free Radic. Biol. Med. 2016, 98, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Dorst, J.; Kuhnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Sheng, L.; Shang, H. Statins and amyotrophic lateral sclerosis: A systematic review and meta-analysis. Amyotroph. Lateral. Scler. Frontotemporal. Degener. 2013, 14, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Bigini, P.; Steffensen, K.R.; Ferrario, A.; Diomede, L.; Ferrara, G.; Barbera, S.; Salzano, S.; Fumagalli, E.; Ghezzi, P.; Mennini, T.; et al. Neuropathologic and biochemical changes during disease progression in liver X receptor beta−/− mice, a model of adult neuron disease. J. Neuropathol. Exp. Neurol. 2010, 69, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.; Gustafsson, N.; Warner, M.; Gustafsson, J.A. Inactivation of liver X receptor beta leads to adultonset motor neuron degeneration in male mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3857–3862. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Fan, X.; Gabbi, C.; Yakimchuk, K.; Parini, P.; Warner, M.; Gustafsson, J.A. Liver X receptor beta (LXRbeta): A link between beta-sitosterol and amyotrophic lateral sclerosis-Parkinson’s dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Mouzat, K.; Molinari, N.; Kantar, J.; Polge, A.; Corcia, P.; Couratier, P.; Clavelou, P.; Juntas-Morales, R.; Pageot, N.; Lobaccaro, J.-A.; et al. Liver X Receptor Genes Variants Modulate ALS Phenotype. Mol. Neurobiol. 2018, 55, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Noh, M.Y.; Kim, H.; Cheon, S.Y.; Lee, K.M.; Lee, J.; Cha, E.; Park, K.S.; Lee, K.W.; Sung, J.J.; et al. 25-Hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 2017, 8, 11855–11867. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2017, 2, e95934. [Google Scholar] [CrossRef] [PubMed]
- Flis, D.J.; Dzik, K.; Kaczor, J.J.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Skeletal Muscle Energy Metabolism, Oxidative Stress, and Mitochondrial Cholesterol Content in Amyotrophic Lateral Sclerosis Mice. Oxid. Med. Cell. Longev. 2018, 2018, 5940748. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Ström, A.L.; Kilty, R.; Venkatakrishnan, P.; White, J.; Everson, W.V.; Smart, E.J.; Zhu, H. Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. FEBS J. 2009, 276, 3308–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milad, N.; White, Z.; Tehrani, A.Y.; Sellers, S.; Rossi, F.M.V.; Bernatchez, P. Increased plasma lipid levels exacerbate muscle pathology in the mdx mouse model of Duchenne muscular dystrophy. Skelet. Muscle 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Radley-Crabb, H.G.; Robertson, T.; Papadimitriou, J.; Spuler, S.; Shavlakadze, T. Lipid accumulation in dysferlin-deficient muscles. Am. J. Pathol. 2014, 184, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova, V.V.; Timonina, N.A.; Zakir’yanova, G.F.; Sokolova, A.V.; Mikhailov, V.M.; Zefirov, A.L.; Krivoi, I.I. The Structural and Functional Characteristics of the Motor End Plates of Dysferlin-Deficient Mice. Neurochem. J. 2018, 12, 305–310. [Google Scholar] [CrossRef]
- Kerr, J.P.; Ward, C.W.; Bloch, R.J. Dysferlin at transverse tubules regulates Ca2+ homeostasis in skeletal muscle. Front. Physiol. 2014, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; Allen, M.V.; Warner, J.L.; Barefield, D.Y.; Krishnan, S.; Swanson, K.E.; Earley, J.U.; McNally, E.M. Enhanced Muscular Dystrophy from Loss of Dysferlin Is Accompanied by Impaired Annexin A6 Translocation after Sarcolemmal Disruption. Am. J. Pathol. 2016, 186, 1610–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, N.; Nonneman, R.J.; Llanga, T.; Dial, C.F.; Riddick, N.V.; Hampton, T.; Moy, S.S.; Lehtimäki, K.K.; Ahtoniemi, T.; Puoliväli, J.; et al. Hip region muscular dystrophy and emergence of motor deficits in dysferlin-deficient Bla/J mice. Physiol. Rep. 2017, 5, e13173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanga, T.; Nagy, N.; Conatser, L.; Dial, C.; Sutton, R.B.; Hirsch, M.L. Structure-Based Designed Nano-Dysferlin Significantly Improves Dysferlinopathy in BLA/J Mice. Mol. Ther. 2017, 25, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Ambery, A.G.; Tackett, L.; Penque, B.A.; Brozinick, J.T.; Elmendorf, J.S. Exercise training prevents skeletal muscle plasma membrane cholesterol accumulation, cortical actin filament loss, and insulin resistance in C57BL/6J mice fed a western-style high-fat diet. Physiol. Rep. 2017, 5, e13363. [Google Scholar] [CrossRef] [PubMed]
- Sellers, S.L.; Milad, N.; White, Z.; Pascoe, C.; Chan, R.; Payne, G.W.; Seow, C.; Rossi, F.; Seidman, M.A.; Bernatchez, P. Increased nonHDL cholesterol levels cause muscle wasting and ambulatory dysfunction in the mouse model of LGMD2B. J. Lipid Res. 2018, 59, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, R.Y.; Nosek, C.M.; Brotto, M.A.; Nishi, M.; Takeshima, H.; Nosek, T.M.; Ma, J. Increased susceptibility to fatigue of slow- and fast-twitch muscles from mice lacking the MG29 gene. Physiol. Genom. 2000, 4, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Brandt, N.R.; Franklin, G.; Brunschwig, J.P.; Caswell, A.H. The role of mitsugumin 29 in transverse tubules of rabbit skeletal muscle. Arch. Biochem. Biophys. 2001, 385, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Brotto, M.A.; Nagaraj, R.Y.; Brotto, L.S.; Takeshima, H.; Ma, J.J.; Nosek, T.M. Defective maintenance of intracellular Ca2+ homeostasis is linked to increased muscle fatigability in the MG29 null mice. Cell Res. 2004, 14, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S.; Berthier, C.; Blaineau, S.; Amsellem, J.; Coronado, R.; Strube, C. Membrane cholesterol modulates dihydropyridine receptor function in mice fetal skeletal muscle cells. J. Physiol. 2004, 555, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, H.; Arner, P.; Gerdhem, P.; Strawbridge, R.J.; Näslund, E.; Thorell, A.; Hamsten, A.; Kere, J.; Dahlman, I. Exome sequencing followed by genotyping suggests SYPL2 as a susceptibility gene for morbid obesity. Eur. J. Hum. Genet. 2015, 23, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 2014, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Brenner, H.R.; Rüegg, M.A. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [PubMed]
- Willadt, S.; Nash, M.; Slater, C.R. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep. 2016, 6, 24849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova, V.V.; Matchkov, V.V.; Bouzinova, E.V.; Vasiliev, A.N.; Razgovorova, I.A.; Heiny, J.A.; Krivoi, I.I. Isoform-specific Na,K-ATPase alterations precede disuse-induced atrophy of rat soleus muscle. BioMed Res. Int. 2015, 2015, 720172. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Karakashian, M.N.; Reid, M.B. Sphingolipid metabolism, oxidant signaling, and contractile function of skeletal muscle. Antioxid. Redox Signal. 2011, 15, 2501–2517. [Google Scholar] [CrossRef] [PubMed]
- Bryndina, I.G.; Shalagina, M.N.; Sekunov, A.V.; Zefirov, A.L.; Petrov, A.M. Clomipramine counteracts lipid raft disturbance due to short-term muscle disuse. Neurosci. Lett. 2018, 664, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Alterman, M.; Dobrowsky, R.T. Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J. Lipid Res. 2005, 46, 1678–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Habegger, K.M.; Hoffman, N.J.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Cerveró, C.; Montull, N.; Tarabal, O.; Piedrafita, L.; Esquerda, J.E.; Calderó, J. Chronic treatment with the AMPK agonist AICAR prevents skeletal muscle pathology but fails to improve clinical outcome in a mouse model of severe spinal muscular atrophy. Neurotherapeutics 2016, 13, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Chibalin, A.V.; Benziane, B.; Zakyrjanova, G.F.; Kravtsova, V.V.; Krivoi, I.I. Early endplate remodeling and skeletal muscle signaling events following rat hindlimb suspension. J. Cell. Physiol. 2018, 233, 6329–6336. [Google Scholar] [CrossRef] [PubMed]
- Vilchinskaya, N.A.; Mochalova, E.P.; Nemirovskaya, T.L.; Mirzoev, T.M.; Turtikova, O.V.; Shenkman, B.S. Rapid decline in MyHC I(β) mRNA expression in rat soleus during hindlimb unloading is associated with AMPK dephosphorylation. J. Physiol. 2017, 595, 7123–7134. [Google Scholar] [CrossRef] [PubMed]
- Vilchinskaya, N.A.; Krivoi, I.I.; Shenkman, B.S. AMP-Activated Protein Kinase as a Key Trigger for the Disuse-Induced Skeletal Muscle Remodeling. Int. J. Mol. Sci. 2018, 19, 3558. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
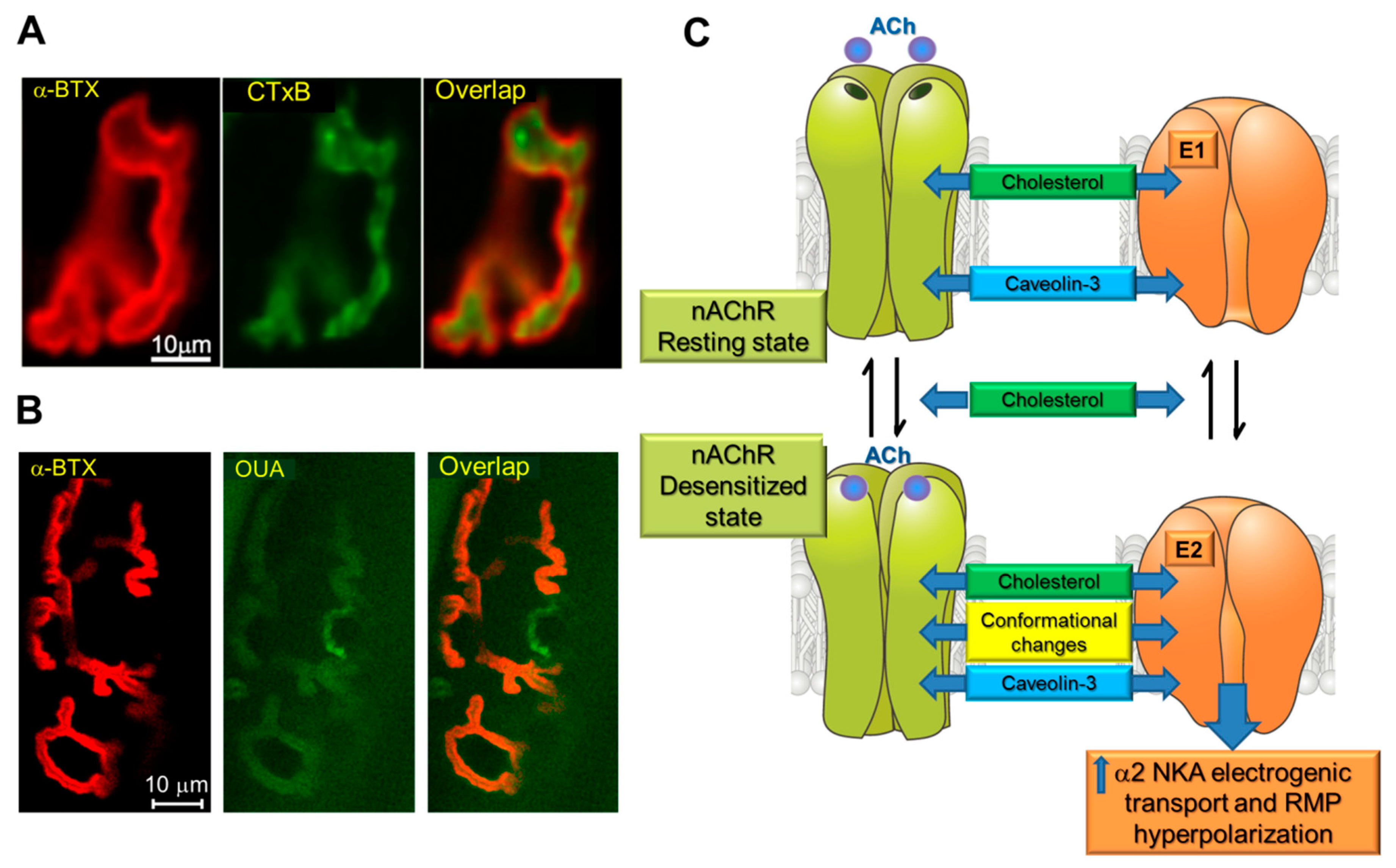{kind=link}
{kind=link}
| Protein | Role in Neuromuscular Transmission | Potential Role of Interaction with Cholesterol | Ref. |
|---|---|---|---|
| P2Y12 receptor | Inhibition of ACh release | Acceleration of the downstream receptor signaling | [84] |
| Ca2+ channels (N, L, P/Q types) | Triggering SV exocytosis in response to AP | Clusterization of channels near exocytotic sites, thereby facilitating exocytosis | [67,68,69] |
| NADPH oxidase (ROS generating enzyme) | Regulation of AP-evoked and spontaneous ACh release | Limitation of background activity | [41,84] |
| Proton pump | Formation of H+ gradient necessary for SV filling with ACh; regulation of SV size | Regulation of precise location and potentiation of H+ transport function | [74,75] |
| Signaling enzymes (protein kinases A/C) | Control of neurotransmitter release | Limitation of background activity of the protein kinases | [42,47] |
| Synapsin | Clusterization of SVs in pools | Lipid raft organization in SV membranes | [81] |
| Synaptophysin | Regulation of exo- and endocytosis | Induction of SV curvature during endocytosis | [52] |
| Synaptotagmin 1 | A major Ca2+ sensor for neurotransmitter release | Location in lipid rafts and precise distribution in presynaptic membrane | [51,70] |
| Syntaxin | Component of SNARE complex mediated SV fusion | Clusterization in membrane; activity-dependent redistribution in lipid rafts | [76,77] |
| Vesicular ACh transporter | Uptake of ACh into SV, nonvesicular ACh release | Precise location in SV membranes and regulation of activity | [36] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivoi, I.I.; Petrov, A.M. Cholesterol and the Safety Factor for Neuromuscular Transmission. Int. J. Mol. Sci. 2019, 20, 1046. https://doi.org/10.3390/ijms20051046
Krivoi II, Petrov AM. Cholesterol and the Safety Factor for Neuromuscular Transmission. International Journal of Molecular Sciences. 2019; 20(5):1046. https://doi.org/10.3390/ijms20051046
Chicago/Turabian StyleKrivoi, Igor I., and Alexey M. Petrov. 2019. "Cholesterol and the Safety Factor for Neuromuscular Transmission" International Journal of Molecular Sciences 20, no. 5: 1046. https://doi.org/10.3390/ijms20051046
APA StyleKrivoi, I. I., & Petrov, A. M. (2019). Cholesterol and the Safety Factor for Neuromuscular Transmission. International Journal of Molecular Sciences, 20(5), 1046. https://doi.org/10.3390/ijms20051046