The Potential Beneficial Effects of Resveratrol on Cardiovascular Complications in Marfan Syndrome Patients–Insights from Rodent-Based Animal Studies


Abstract
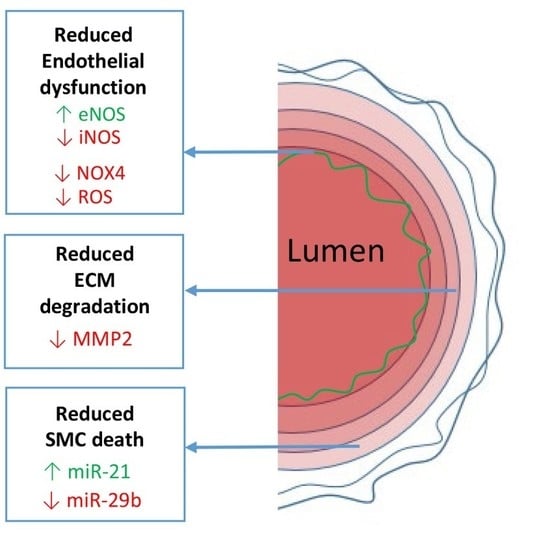
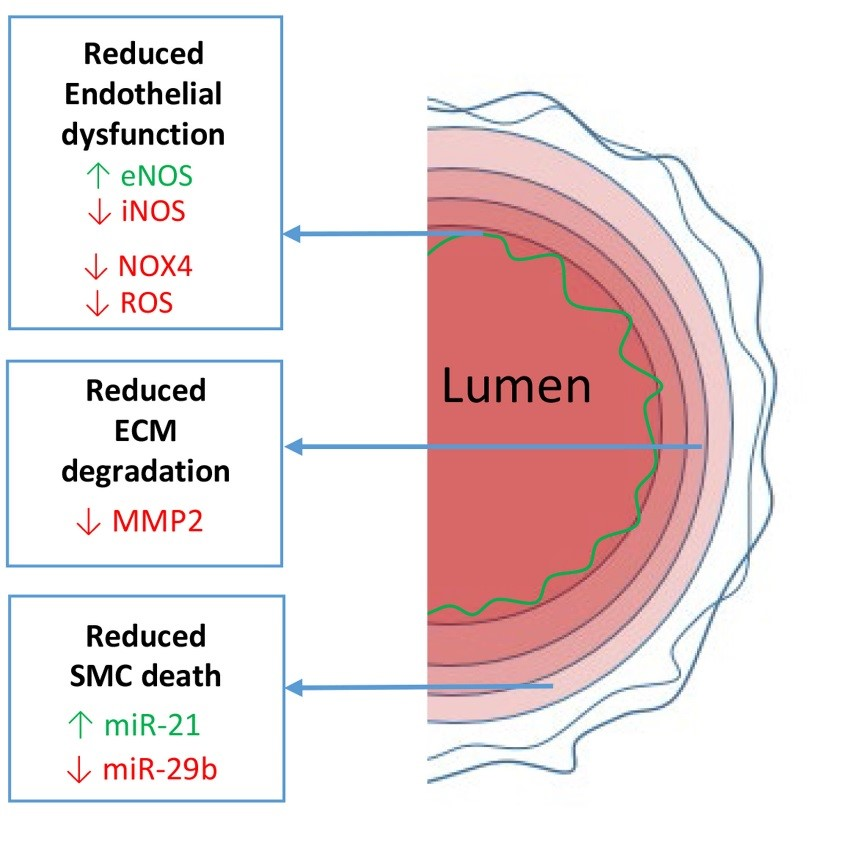
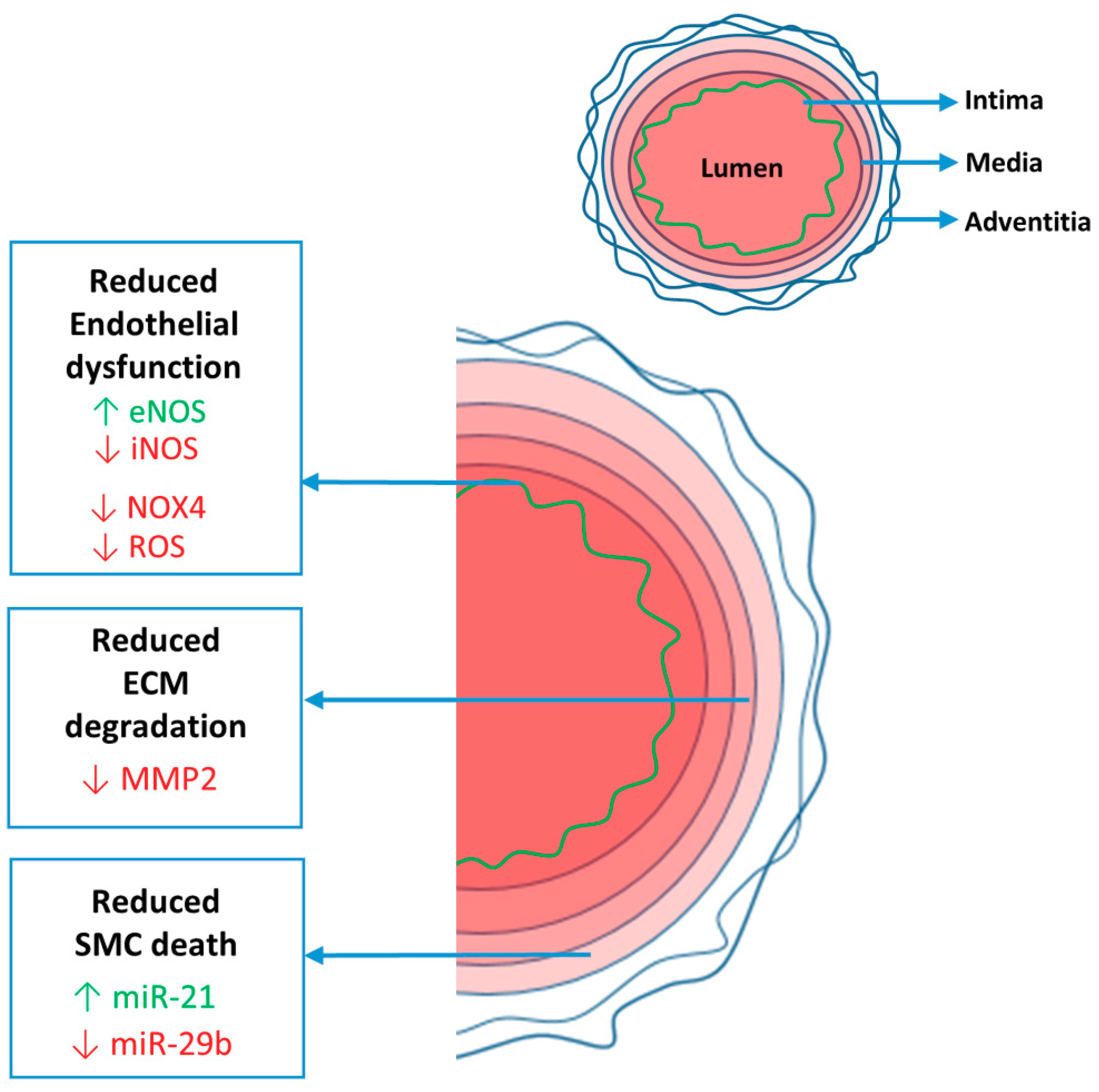
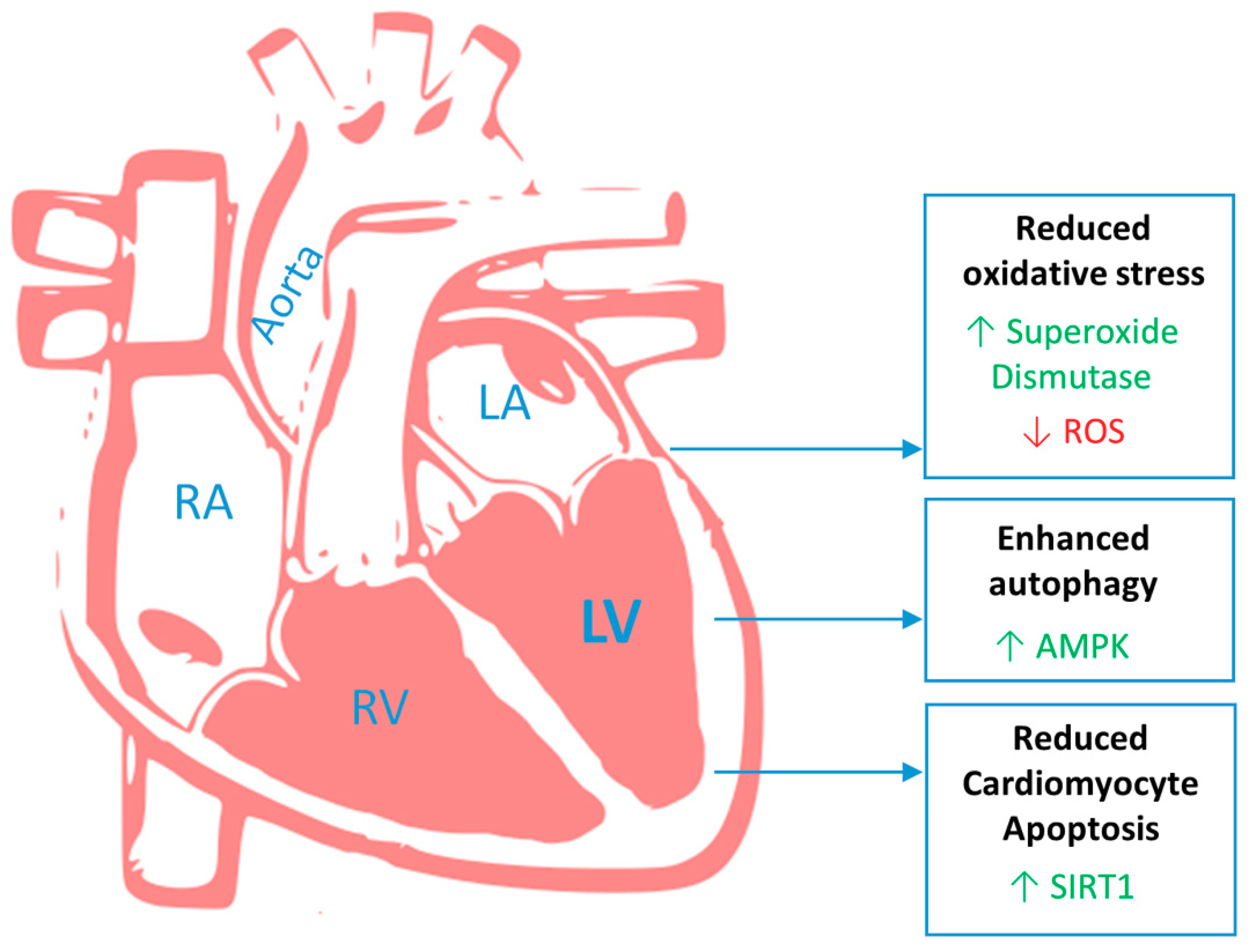
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Aorta
2.1. Aortic Aneurysm
2.2. Endothelial Dysfunction
2.2.1. eNOS/iNOS Balance
2.2.2. NOX4 and ROS
2.3. Medial Degeneration
2.3.1. ECM Degeneration
2.3.2. SMC Death
3. Heart
3.1. Mitral Valve Prolapse
3.2. Left Ventricular Impairment
4. Exercise Mimetic
5. Conclusion
Funding
Conflicts of Interest
Abbreviations
| AAA | Abdominal aortic aneurysm |
| ACE2 | Angiotensin-converting enzyme 2 |
| AGTR1 | Angiotensin type-1 receptor (Agtr1 [mouse]) |
| AMPK | AMP-activated protein kinase |
| ARB | Angiotensin II receptor blocker |
| ECM | Extracellular matrix |
| eNOS | Endothelial nitric oxide synthase |
| FBN1 | Fibrillin-1 (Fbn1 [mouse]) |
| iNOS | Inducible nitric oxide synthase |
| MFS | Marfan syndrome |
| MMP | Matrix metalloproteinase |
| NFκB | Nuclear factor kappa light chain enhancer of activated B cells |
| PGC1a | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| RES | Resveratrol |
| ROS | Reactive oxygen species |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| SMC | Smooth muscle cell |
| TGF-β | Transforming growth factor beta |
References
- Silverman, D.I.; Burton, K.J.; Gray, J.; Bosner, M.S.; Kouchoukos, N.T.; Roman, M.J.; Boxer, M.; Devereux, R.B.; Tsipouras, P. Life expectancy in the Marfan syndrome. Am. J. Cardiol. 1995, 75, 157–160. [Google Scholar] [CrossRef]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar] [CrossRef]
- den Hartog, A.W.; Franken, R.; Zwinderman, A.H.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; de Waard, V.; Pals, G.; Mulder, B.J.; Groenink, M. The risk for type B aortic dissection in Marfan syndrome. J. Am. Coll. Cardiol. 2015, 65, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Teixido-Tura, G.; Forteza, A.; Rodriguez-Palomares, J.; Gonzalez Mirelis, J.; Gutierrez, L.; Sanchez, V.; Ibanez, B.; Garcia-Dorado, D.; Evangelista, A. Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome. J. Am. Coll. Cardiol. 2018, 72, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Ramirez, F. Therapies for Thoracic Aortic Aneurysms and Acute Aortic Dissections. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Collod-Beroud, G.; Le Bourdelles, S.; Ades, L.; Ala-Kokko, L.; Booms, P.; Boxer, M.; Child, A.; Comeglio, P.; De Paepe, A.; Hyland, J.C.; et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum. Mutat. 2003, 22, 199–208. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Loeys, B.; Leroy, B.; Coucke, P.; Dietz, H.; De Paepe, A. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin. Genet. 2007, 72, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Hibender, S.; Franken, R.; van Roomen, C.; Ter Braake, A.; van der Made, I.; Schermer, E.E.; Gunst, Q.; van den Hoff, M.J.; Lutgens, E.; Pinto, Y.M.; et al. Resveratrol Inhibits Aortic Root Dilatation in the Fbn1C1039G/+ Marfan Mouse Model. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Dyck, J.R. Therapeutic potential of resveratrol in heart failure. Ann. N. Y. Acad. Sci. 2015, 1348, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont-Rousselot, D. Resveratrol and Cardiovascular Diseases. Nutrients 2016, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Anzai, T.; Morisawa, M.; Kohno, T.; Nagai, T.; Anzai, A.; Takahashi, T.; Shimoda, M.; Sasaki, A.; Maekawa, Y.; et al. Resveratrol prevents the development of abdominal aortic aneurysm through attenuation of inflammation, oxidative stress, and neovascularization. Atherosclerosis 2011, 217, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Pane, B.; Barisione, C.; Spinella, G.; Garibaldi, S.; Ghigliotti, G.; Brunelli, C.; Fulcheri, E.; Palombo, D. Resveratrol counteracts systemic and local inflammation involved in early abdominal aortic aneurysm development. J. Surg. Res. 2011, 171, e237–e246. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.S.; Biros, E.; Krishna, S.M.; Wang, Y.; Tikellis, C.; Morton, S.K.; Moxon, J.V.; Cooper, M.E.; Norman, P.E.; Burrell, L.M.; et al. Resveratrol Inhibits Growth of Experimental Abdominal Aortic Aneurysm Associated With Upregulation of Angiotensin-Converting Enzyme 2. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2195–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeman, J.H.; Ashcroft, B.A.; Beenakker, J.W.; van Es, M.; Koekkoek, N.B.; Prins, F.A.; Tielemans, J.F.; Abdul-Hussien, H.; Bank, R.A.; Oosterkamp, T.H. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 862–865. [Google Scholar] [PubMed]
- Adams, J.N.; Trent, R.J. Aortic complications of Marfan’s syndrome. Lancet 1998, 352, 1722–1723. [Google Scholar] [CrossRef]
- Hartog, A.W.; Franken, R.; Zwinderman, A.H.; Groenink, M.; Mulder, B.J. Current and future pharmacological treatment strategies with regard to aortic disease in Marfan syndrome. Expert Opin Pharmacother, Expert Opin. Pharmacother. 2012, 13, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Groenink, M.; den Hartog, A.W.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Andrikopoulos, K.; Tian, J.; Lee, S.Y.; Keene, D.R.; Ono, R.; Reinhardt, D.P.; Sakai, L.Y.; Biery, N.J.; Bunton, T.; et al. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet. 1997, 17, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, A.J.; Wilson, N.K.; Habashi, J.P.; Bedja, D.; Whitworth, R.E.; Lindsay, M.E.; Schoenhoff, F.; Myers, L.; Huso, N.; et al. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife 2015, 4, e08648. [Google Scholar] [CrossRef] [PubMed]
- Wanga, S.; Hibender, S.; Ridwan, Y.; van Roomen, C.; Vos, M.; van der Made, I.; van Vliet, N.; Franken, R.; van Riel, L.A.; Groenink, M.; et al. Aortic microcalcification is associated with elastin fragmentation in Marfan syndrome. J. Pathol. 2017, 243, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ. Res. 2011, 109, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Attia, R.; Mayr, U.; Gomes, R.S.; Phinikaridou, A.; Yin, X.; Langley, S.R.; Willeit, P.; Lu, R.; Fanshawe, B.; et al. Role of miR-195 in aortic aneurysmal disease. Circ. Res. 2014, 115, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.R.; Chin, J.T.; Dake, B.A.; Maegdefessel, L.; Miller, M.O.; Kimura, N.; Tsao, P.S.; Iosef, C.; Berry, G.J.; Mohr, F.W.; et al. miR-29b participates in early aneurysm development in Marfan syndrome. Circ. Res. 2012, 110, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Deng, A.; Merk, D.R.; Raiesdana, A.; Leeper, N.J.; Raaz, U.; Schoelmerich, A.M.; McConnell, M.V.; et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci. Transl. Med. 2012, 4, 122ra22. [Google Scholar] [CrossRef] [PubMed]
- Radonic, T.; de Witte, P.; Groenink, M.; de Waard, V.; Lutter, R.; van Eijk, M.; Jansen, M.; Timmermans, J.; Kempers, M.; Scholte, A.J.; et al. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS ONE 2012, 7, e32963. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Hibender, S.; den Hartog, A.W.; Radonic, T.; de Vries, C.J.; Zwinderman, A.H.; Groenink, M.; Mulder, B.J.; de Waard, V. No beneficial effect of general and specific anti-inflammatory therapies on aortic dilatation in Marfan mice. PLoS ONE 2014, 9, e107221. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Amiya, E.; Watanabe, M.; Omori, K.; Imai, Y.; Fujita, D.; Nishimura, H.; Kato, M.; Morota, T.; Nawata, K.; et al. Impairment of flow-mediated dilation correlates with aortic dilation in patients with Marfan syndrome. Heart Vessels 2014, 29, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Au Yeung, K.; Cortes, S.F.; Sandor, G.G.; Judge, D.P.; Dietz, H.C.; van Breemen, C. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br. J. Pharmacol. 2007, 150, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Buluc, M.; Demirel-Yilmaz, E. Resveratrol decreases calcium sensitivity of vascular smooth muscle and enhances cytosolic calcium increase in endothelium. Vascul. Pharmacol. 2006, 44, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.E.; Soria-Castro, E.; Lans, V.G.; Ontiveros, E.M.; Mejia, B.I.; Hernandez, H.J.; Garcia, R.B.; Herrera, V.; Perez-Torres, I. Analysis of oxidative stress enzymes and structural and functional proteins on human aortic tissue from different aortopathies. Oxid. Med. Cell. Longev. 2014, 2014, 760694. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, O.; Perez-Torres, I.; Marquez, R.; Criales, S.; Mejia, A.M.; Chiney, C.; Hernandez-Lemus, E.; Soto, M.E. The Evaluation of Flow-Mediated Vasodilation in the Brachial Artery Correlates With Endothelial Dysfunction Evaluated by Nitric Oxide Synthase Metabolites in Marfan Syndrome Patients. Front. Physiol. 2018, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Oller, J.; Mendez-Barbero, N.; Ruiz, E.J.; Villahoz, S.; Renard, M.; Canelas, L.I.; Briones, A.M.; Alberca, R.; Lozano-Vidal, N.; Hurle, M.A.; et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med. 2017, 23, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.; Gongol, B.; Chen, Z.; Woo, B.; Subramaniam, S.; Chien, S.; Shyy, J.Y. Mechanosensitive microRNAs-role in endothelial responses to shear stress and redox state. Free Radic. Biol. Med. 2013, 64, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Fledderus, J.O.; Volger, O.L.; van Wanrooij, E.J.; Pardali, E.; Weesie, F.; Kuiper, J.; Pannekoek, H.; ten Dijke, P.; Horrevoets, A.J. KLF2 suppresses TGF-beta signaling in endothelium through induction of Smad7 and inhibition of AP-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; den Hartog, A.W.; de Waard, V.; Engele, L.; Radonic, T.; Lutter, R.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Zwinderman, A.H.; et al. Circulating transforming growth factor-beta as a prognostic biomarker in Marfan syndrome. Int. J. Cardiol. 2013, 168, 2441–2446. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Villarreal, G., Jr.; Zhang, Y.; Garcia-Cardena, G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc. Res. 2010, 85, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Wallerath, T.; Deckert, G.; Ternes, T.; Anderson, H.; Li, H.; Witte, K.; Forstermann, U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation 2002, 106, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Leikert, J.F.; Rathel, T.R.; Wohlfart, P.; Cheynier, V.; Vollmar, A.M.; Dirsch, V.M. Red wine polyphenols enhance endothelial nitric oxide synthase expression and subsequent nitric oxide release from endothelial cells. Circulation 2002, 106, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Chakrabarti, S.; Pereira, T.J.; Oka, T.; Levasseur, J.; Beker, D.; Zordoky, B.N.; Morton, J.S.; Nagendran, J.; Lopaschuk, G.D.; et al. Resveratrol prevents hypertension and cardiac hypertrophy in hypertensive rats and mice. Biochim. Biophys. Acta 2013, 1832, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.; Baker, M.B.; Moore, J.P.; Searles, C.D. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem. Biophys. Res. Commun. 2010, 393, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.H.; van Breemen, C.; Chung, A.W. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascul. Pharmacol. 2010, 52, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.E.; Iturriaga Hernandez, A.V.; Guarner Lans, V.; Zuniga-Munoz, A.; Aranda Fraustro, A.; Velazquez Espejel, R.; Perez-Torres, I. Participation of oleic acid in the formation of the aortic aneurysm in Marfan syndrome patients. Prostag. Other Lipid Mediat. 2016, 123, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, G.; Heemskerk, N.; van Buul, J.D.; de Waard, V. The Ins and Outs of Small GTPase Rac1 in the Vasculature. J. Pharmacol. Exp. Ther. 2015, 354, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarzuelo, M.J.; Lopez-Sepulveda, R.; Sanchez, M.; Romero, M.; Gomez-Guzman, M.; Ungvary, Z.; Perez-Vizcaino, F.; Jimenez, R.; Duarte, J. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochem. Pharmacol. 2013, 85, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Altayo, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Del Blanco, D.G.; Lopez-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barbera, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. Elastin in large artery stiffness and hypertension. J. Cardiovasc. Transl. Res. 2012, 5, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Milewicz Dianna, M.; Trybus Kathleen, M.; Guo, D.-c.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull James, T. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Backer, J. Cardiovascular characteristics in Marfan syndrome and their relation to the genotype. Verh. K. Acad. Geneeskd. Belg. 2009, 71, 335–371. [Google Scholar] [PubMed]
- Nataatmadja, M.; West, M.; West, J.; Summers, K.; Walker, P.; Nagata, M.; Watanabe, T. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation 2003, 108 (Suppl. 1), Ii329–Ii334. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Davlouros, P.A.; McCarthy, K.P.; Gatzoulis, M.A.; Ho, S.Y. Intrinsic histological abnormalities of aortic root and ascending aorta in tetralogy of Fallot: evidence of causative mechanism for aortic dilatation and aortopathy. Int. J. Cardiol. 2019, 278, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Lehsau, A.C.; Sauer, B.; Pieper, B.; Mohamed, S.A. Comparison of biomechanical properties in ascending aortic aneurysms of patients with congenital bicuspid aortic valve and Marfan syndrome. Int. J. Cardiol. 2019, 278, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, F.; Mazzaglia, D.; Pellegrino, A.; Grego, S.; Fiorito, R.; Ferlosio, A.; Chiariello, L.; Orlandi, A. Aortopathy in Marfan syndrome: an update. Cardiovasc. Pathol. 2014, 23, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Fedak, P.W.; de Sa, M.P.; Verma, S.; Nili, N.; Kazemian, P.; Butany, J.; Strauss, B.H.; Weisel, R.D.; David, T.E. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J. Thorac. Cardiovasc. Surg. 2003, 126, 797–806. [Google Scholar] [CrossRef]
- Xiong, W.; Meisinger, T.; Knispel, R.; Worth, J.M.; Baxter, B.T. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ. Res. 2012, 110, e92–e101. [Google Scholar] [CrossRef] [PubMed]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Pluijm, I.; Burger, J.; van Heijningen, P.M.; IJpma, A.; van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef] [PubMed]
- Meijer, C.A.; Stijnen, T.; Wasser, M.N.; Hamming, J.F.; van Bockel, J.H.; Lindeman, J.H. Doxycycline for stabilization of abdominal aortic aneurysms: a randomized trial. Ann. Intern. Med. 2013, 159, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45 (Suppl. A), A25–A32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Grewal, N.; Franken, R.; Mulder, B.J.; Goumans, M.J.; Lindeman, J.H.; Jongbloed, M.R.; DeRuiter, M.C.; Klautz, R.J.; Bogers, A.J.; Poelmann, R.E.; et al. Histopathology of aortic complications in bicuspid aortic valve versus Marfan syndrome: relevance for therapy? Heart Vessels 2016, 31, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Dale, M.; Fitzgerald, M.P.; Liu, Z.; Meisinger, T.; Karpisek, A.; Purcell, L.N.; Carson, J.S.; Harding, P.; Lang, H.; Koutakis, P.; et al. Premature aortic smooth muscle cell differentiation contributes to matrix dysregulation in Marfan Syndrome. PLoS ONE 2017, 12, e0186603. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Meirelles, T.; Lopez-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko Del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizabal, Y.; et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Deuse, T.; Stubbendorff, M.; Chernogubova, E.; Erben, R.G.; Eken, S.M.; Jin, H.; Li, Y.; Busch, A.; Heeger, C.H.; et al. Local MicroRNA Modulation Using a Novel Anti-miR-21-Eluting Stent Effectively Prevents Experimental In-Stent Restenosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Aalberts, J.J.; van Tintelen, J.P.; Meijboom, L.J.; Polko, A.; Jongbloed, J.D.; van der Wal, H.; Pals, G.; Osinga, J.; Timmermans, J.; de Backer, J.; et al. Relation between genotype and left-ventricular dilatation in patients with Marfan syndrome. Gene 2014, 534, 40–43. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Loeys, B.; Devos, D.; Dietz, H.; De Sutter, J.; De Paepe, A. A critical analysis of minor cardiovascular criteria in the diagnostic evaluation of patients with Marfan syndrome. Genet. Med. 2006, 8, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyeritz, R.E.; Wappel, M.A. Mitral valve dysfunction in the Marfan syndrome.; Clinical and echocardiographic study of prevalence and natural history. Am. J. Med. 1983, 74, 797–807. [Google Scholar] [CrossRef]
- Detaint, D.; Faivre, L.; Collod-Beroud, G.; Child, A.H.; Loeys, B.L.; Binquet, C.; Gautier, E.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur. Heart J. 2010, 31, 2223–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avierinos, J.F.; Gersh, B.J.; Melton, L.J.; Bailey, K.R.; Shub, C.; Nishimura, R.A.; Tajik, A.J.; Enriquez-Sarano, M. Natural history of asymptomatic mitral valve prolapse in the community. Circulation 2002, 106, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M.; Cheng, A.; Myers, L.A.; Martinez-Murillo, F.; Jie, C.; Bedja, D.; Gabrielson, K.L.; Hausladen, J.M.; Mecham, R.P.; Judge, D.P.; et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Investig. 2004, 114, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Tae, H.J.; Petrashevskaya, N.; Marshall, S.; Krawczyk, M.; Talan, M. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H290–H299. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.F.; Devos, D.; Segers, P.; Matthys, D.; Francois, K.; Gillebert, T.C.; De Paepe, A.M.; De Sutter, J. Primary impairment of left ventricular function in Marfan syndrome. Int. J. Cardiol. 2006, 112, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.R.; Carta, L.; Benard, L.; Chemaly, E.R.; Chiu, E.; Rao, S.K.; Hampton, T.G.; Yurchenco, P.; Costa, K.D.; Hajjar, R.J.; et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J. Clin. Investig. 2014, 124, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steijns, F.; van Hengel, J.; Sips, P.; De Backer, J.; Renard, M. A heart for fibrillin: spatial arrangement in adult wild-type murine myocardial tissue. Histochem. Cell Biol. 2018, 150, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Campens, L.; Renard, M.; Trachet, B.; Segers, P.; Muino Mosquera, L.; De Sutter, J.; Sakai, L.; De Paepe, A.; De Backer, J. Intrinsic cardiomyopathy in Marfan syndrome: results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr. Res. 2015, 78, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Fatima, N.; Zaman, M.U.; Hashmi, A.; Kamal, S.; Hameed, A. Assessing adriamycin-induced early cardiotoxicity by estimating left ventricular ejection fraction using technetium-99m multiple-gated acquisition scan and echocardiography. Nucl. Med. Commun. 2011, 32, 381–385. [Google Scholar] [CrossRef] [PubMed]
- McKillop, J.H.; Bristow, M.R.; Goris, M.L.; Billingham, M.E.; Bockemuehl, K. Sensitivity and specificity of radionuclide ejection fractions in doxorubicin cardiotoxicity. Am. Heart J. 1983, 106 Pt 1, 1048–1056. [Google Scholar] [CrossRef]
- Scott, J.M.; Khakoo, A.; Mackey, J.R.; Haykowsky, M.J.; Douglas, P.S.; Jones, L.W. Modulation of anthracycline-induced cardiotoxicity by aerobic exercise in breast cancer: current evidence and underlying mechanisms. Circulation 2011, 124, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, Y.; Qu, S.; Wei, X.; Zhu, H.; Luo, Q.; Liu, M.; Chen, G.; Xiao, X. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through SIRT1-mediated deacetylation of p53. Cardiovasc. Res. 2011, 90, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Song, Z.P.; Gui, D.M.; Hu, W.; Chen, Y.G.; Zhang, D.D. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in lymphoma nude mice by heme oxygenase-1 induction. Cardiovasc. Toxicol. 2012, 12, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Sin, T.K.; Tam, B.T.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Ying, M.; Rudd, J.A.; Siu, P.M. Resveratrol protects against doxorubicin-induced cardiotoxicity in aged hearts through the SIRT1-USP7 axis. J. Physiol. 2015, 593, 1887–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.; Dong, C.; Patel, J.; Duan, C.; Wang, X.; Wu, X.; Cao, Y.; Pu, L.; Lu, D.; Shen, T.; et al. SIRT1 suppresses doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell. Physiol. Biochem. 2015, 35, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hu, W.; Song, Z.P.; Chen, Y.G.; Zhang, D.D.; Wang, C.Q. Resveratrol-induced autophagy promotes survival and attenuates doxorubicin-induced cardiotoxicity. Int. Immunopharmacol. 2016, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chen, Y.; Liao, L.; Wu, W. Resveratrol protects HUVECs from oxidized-LDL induced oxidative damage by autophagy upregulation via the AMPK/SIRT1 pathway. Cardiovasc. Drugs Ther. 2013, 27, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chang, H.; Fu, Y.J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Huang, Y.; Zheng, W.; Yan, J.; Cheng, M.; Zhao, R.; Chen, L.; Hu, C.; Jia, W. Resveratrol reduces intracellular reactive oxygen species levels by inducing autophagy through the AMPK-mTOR pathway. Front. Med. 2018, 12, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, D.; Moon, H.Y.; van Praag, H. Exercise in a Pill: The Latest on Exercise-Mimetics. Brain Plast 2017, 2, 153–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliemann, L.; Nyberg, M.; Hellsten, Y. Effects of exercise training and resveratrol on vascular health in aging. Free Radic. Biol. Med. 2016, 98, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Mas-Stachurska, A.; Siegert, A.M.; Batlle, M.; Gorbenko Del Blanco, D.; Meirelles, T.; Rubies, C.; Bonorino, F.; Serra-Peinado, C.; Bijnens, B.; Baudin, J.; et al. Cardiovascular Benefits of Moderate Exercise Training in Marfan Syndrome: Insights From an Animal Model. J. Am. Heart Assoc. 2017, 6, e006438. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.; Nielsen, C.; Alex, R.; Cooper, K.; Farney, M.; Gaufin, D.; Cui, J.Z.; van Breemen, C.; Broderick, T.L.; Vallejo-Elias, J.; et al. Mild aerobic exercise blocks elastin fiber fragmentation and aortic dilatation in a mouse model of Marfan syndrome associated aortic aneurysm. J. Appl. Physiol. 2017, 123, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Itoga, N.K.; Rothenberg, K.A.; Suarez, P.; Ho, T.V.; Mell, M.W.; Xu, B.; Curtin, C.M.; Dalman, R.L. Metformin prescription status and abdominal aortic aneurysm disease progression in the U.S. veteran population. J. Vasc. Surg. 2018, 67, e52. [Google Scholar] [CrossRef]
- Fogacci, F.; Tocci, G.; Presta, V.; Fratter, A.; Borghi, C.; Cicero, A.F.G. Effect of resveratrol on blood pressure: A systematic review and meta-analysis of randomized, controlled, clinical trials. Crit. Rev. Food Sci. Nutr. 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Andel, M.M.; Groenink, M.; Zwinderman, A.H.; Mulder, B.J.M.; de Waard, V. The Potential Beneficial Effects of Resveratrol on Cardiovascular Complications in Marfan Syndrome Patients–Insights from Rodent-Based Animal Studies. Int. J. Mol. Sci. 2019, 20, 1122. https://doi.org/10.3390/ijms20051122
van Andel MM, Groenink M, Zwinderman AH, Mulder BJM, de Waard V. The Potential Beneficial Effects of Resveratrol on Cardiovascular Complications in Marfan Syndrome Patients–Insights from Rodent-Based Animal Studies. International Journal of Molecular Sciences. 2019; 20(5):1122. https://doi.org/10.3390/ijms20051122
Chicago/Turabian Stylevan Andel, Mitzi M., Maarten Groenink, Aeilko H. Zwinderman, Barbara J.M. Mulder, and Vivian de Waard. 2019. "The Potential Beneficial Effects of Resveratrol on Cardiovascular Complications in Marfan Syndrome Patients–Insights from Rodent-Based Animal Studies" International Journal of Molecular Sciences 20, no. 5: 1122. https://doi.org/10.3390/ijms20051122
APA Stylevan Andel, M. M., Groenink, M., Zwinderman, A. H., Mulder, B. J. M., & de Waard, V. (2019). The Potential Beneficial Effects of Resveratrol on Cardiovascular Complications in Marfan Syndrome Patients–Insights from Rodent-Based Animal Studies. International Journal of Molecular Sciences, 20(5), 1122. https://doi.org/10.3390/ijms20051122