Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients



{kind=link}
Abstract
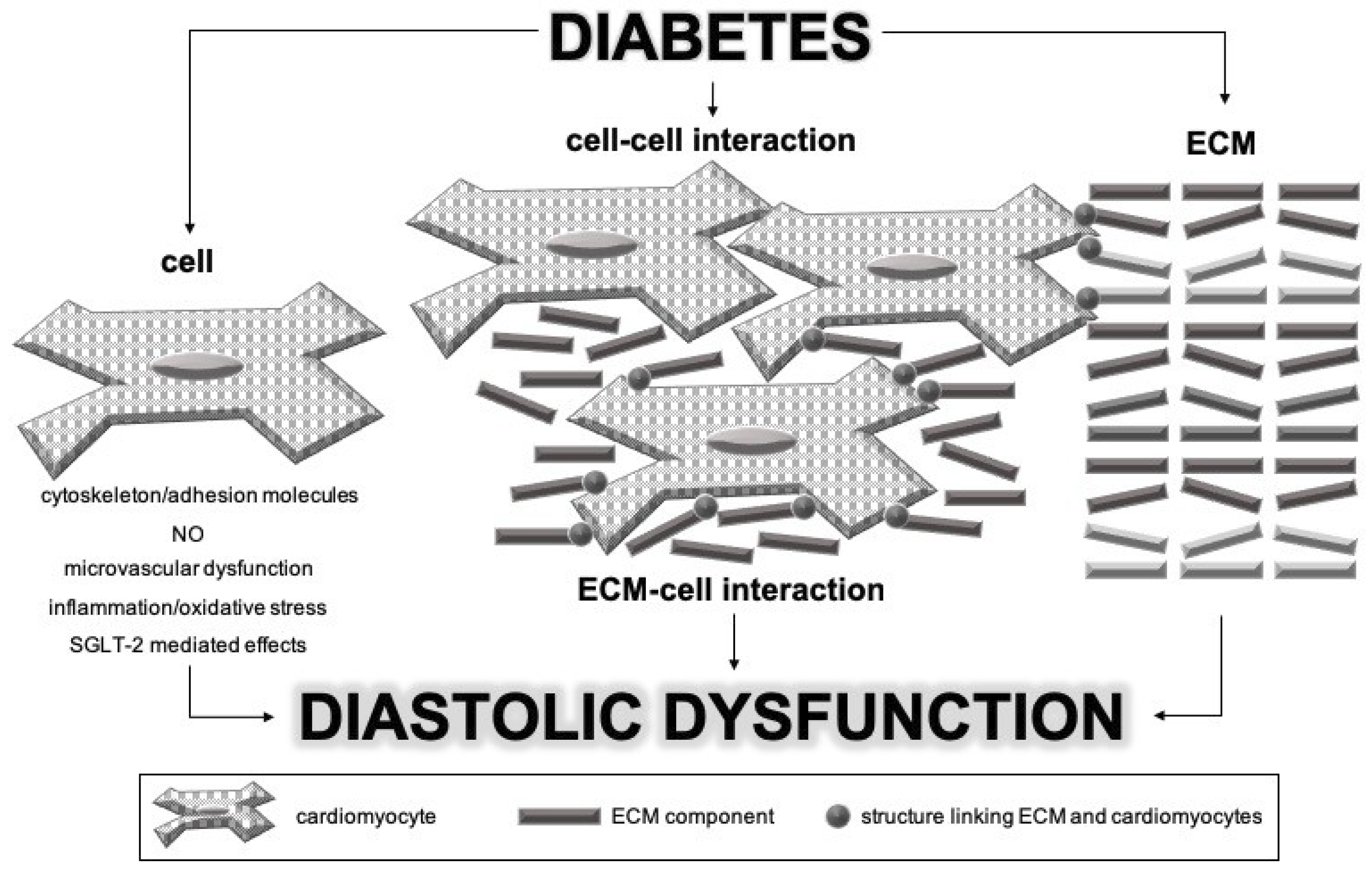
:1. Introduction
2. Pathophysiological Process Underlying Diabetic Cardiomyopathy
3. Models of Diabetic Cardiomyopathy
4. Diabetes Mellitus and Increased Intrinsic Cardiomyocyte Cell Stiffness
5. Nitric Oxide and Increased Contractile State of Cardiomyocytes in Diabetes
6. Inflammation and Oxidative Stress in Cardiomyocytes in Diabetes
7. Coronary Microvascular Dysfunction in Diabetes
8. Various Transporters and Substrate Metabolism in Cardiomyocytes in Diabetes
9. Sodium Glucose Cotransporter-2-Mediated Effects in Cardiomyocytes in Diabetes
10. Extracellular Matrix under Normal Physiology and in Pathophysiological States in Diabetic Myocardium
11. Interactions between Cells and Extracellular Matrix in Diabetic Myocardium
12. Potential Targets to Prevent and Treat Diastolic Dysfunction in Diabetes
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | Angiotensin converting enzyme |
| AGE | Advanced glycation end product |
| ARNi | Angiotensin receptor-neprilysin inhibitor |
| ATM | Atomic force microscopy |
| ATP | Adenosine 5′-triphosphate |
| cGMP | Cyclic guanosine monophosphate |
| CIRKO | Cardiomyocyte selective insulin receptor knockout mice |
| CMRI | Cardiac magnetic resonance imaging |
| DCM | Diabetic cardiomyopathy |
| DM | Diabetes mellitus |
| e/n/iNOS | Endothelial/neuronal/inducible NO synthase |
| ECM | Extracellular matrix |
| FABP | Fatty acid-binding protein |
| FATP | Fatty acid transport protein |
| FFA | Free fatty acid |
| FMD | Flow mediated dilation |
| GK | Goto-Kakizaki rats |
| GLP-1RA | Glucagon like peptide-1 receptor antagonist |
| GLUT | Facilitative glucose transporters |
| HFD | High fat diet |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| HNE | Na+/H+ exchanger |
| ICAM-1 | Intercellular cell adhesion molecule-1 |
| KB | Ketone body |
| MACE | Major adverse cardiovascular events |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MHC-PPARα | Mice with cardiomyocyte specific overexpression of the transcription factor for peroxisome proliferator activated receptor α |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NCX | Na+/Ca2+ exchanger |
| NF-κB | Nuclear transcription factor-κB |
| NO | Nitric oxide |
| NOD | Non-obese diabetic mice |
| NP | Natriuretic peptide |
| NT-proBNP | N-terminal prohormone of brain natriuretic peptide |
| PET | Positron emission tomography |
| RAAS | Renin-angiotensin-aldosterone system |
| ROS | Reactive oxygen species |
| RT | Resting tension |
| SERCA2 | Sarco/endoplasmic reticulum Ca2+-ATPase 2 |
| SGK1 | Serum and glucocorticoid-regulated kinase-1 |
| SGLT | Sodium glucose cotransporter |
| SR | Sarcoplasmic reticulum |
| STZ | Streptozotocin |
| TCA | Tricarboxylic acid |
| TDI | Tissue doppler imaging |
| TGFβ | Transforming growth factor β |
| TNFα | Tumor necrosis factor α |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| ZDF | Zucker diabetic fatty rats |
References
- Boonman-de Winter, L.J.; Rutten, F.H.; Cramer, M.J.; Landman, M.J.; Liem, A.H.; Rutten, G.E.; Hoes, A.W. High prevalence of previously unknown heart failure and left ventricular dysfunction in patients with type 2 diabetes. Diabetologia 2012, 55, 2154–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrainsdottir, I.S.; Aspelund, T.; Thorgeirsson, G.; Gudnason, V.; Hardarson, T.; Malmberg, K.; Sigurdsson, G.; Ryden, L. The association between glucose abnormalities and heart failure in the population-based reykjavik study. Diabetes Care 2005, 28, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Kengne, A.P.; Turnbull, F.; MacMahon, S. The framingham study, diabetes mellitus and cardiovascular disease: Turning back the clock. Prog. Cardiovasc. Dis. 2010, 53, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Deedwania, P.C.; Giles, T.D.; Klibaner, M.; Ghali, J.K.; Herlitz, J.; Hildebrandt, P.; Kjekshus, J.; Spinar, J.; Vitovec, J.; Stanbrook, H.; et al. Efficacy, safety and tolerability of metoprolol CR/XL in patients with diabetes and chronic heart failure: Experiences from MERIT-HF. Am. Heart J. 2005, 149, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Vaur, L.; Gueret, P.; Lievre, M.; Chabaud, S.; Passa, P.; study, D.S.G. Development of congestive heart failure in type 2 diabetic patients with microalbuminuria or proteinuria: Observations from the DIABHYCAR (type 2 DIAbetes, Hypertension, CArdiovascular Events and Ramipril) study. Diabetes Care 2003, 26, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Ryden, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.P.; et al. ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The task force on diabetes, pre-diabetes, and cardiovascular diseases of the european society of cardiology (ESC) and developed in collaboration with the european association for the study of diabetes (EASD). Eur. Heart J. 2013, 34, 3035–3087. [Google Scholar]
- Dandamudi, S.; Slusser, J.; Mahoney, D.W.; Redfield, M.M.; Rodeheffer, R.J.; Chen, H.H. The prevalence of diabetic cardiomyopathy: A population-based study in olmsted county, minnesota. J. Card. Fail. 2014, 20, 304–309. [Google Scholar] [CrossRef]
- Schannwell, C.M.; Schneppenheim, M.; Perings, S.; Plehn, G.; Strauer, B.E. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology 2002, 98, 33–39. [Google Scholar] [CrossRef]
- Shimizu, M.; Umeda, K.; Sugihara, N.; Yoshio, H.; Ino, H.; Takeda, R.; Okada, Y.; Nakanishi, I. Collagen remodelling in myocardia of patients with diabetes. J. Clin. Pathol. 1993, 46, 32–36. [Google Scholar] [CrossRef]
- Eguchi, K.; Boden-Albala, B.; Jin, Z.; Rundek, T.; Sacco, R.L.; Homma, S.; di Tullio, M.R. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am. J. Cardiol. 2008, 101, 1787–1791. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Franssen, C.P.; Hamdani, N.; Verheugt, F.W.; Somsen, G.A.; Paulus, W.J. Molecular and cellular basis for diastolic dysfunction. Curr. Heart Fail. Rep. 2012, 9, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Galderisi, M. Diastolic dysfunction and diastolic heart failure: Diagnostic, prognostic and therapeutic aspects. Cardiovasc. Ultrasound 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, I.; Macedo, R.; Bluemke, D.A.; Lima, J.A. Magnetic resonance imaging in the evaluation of non-ischemic cardiomyopathies: Current applications and future perspectives. Heart Fail. Rev. 2006, 11, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Depre, C.; Young, M.E.; Ying, J.; Ahuja, H.S.; Han, Q.; Garza, N.; Davies, P.J.; Taegtmeyer, H. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J. Mol. Cell. Cardiol. 2000, 32, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Sun, Y.; Tyagi, S.C.; Cleutjens, J.P. Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 1994, 26, 279–292. [Google Scholar] [CrossRef]
- Fomovsky, G.M.; Thomopoulos, S.; Holmes, J.W. Contribution of extracellular matrix to the mechanical properties of the heart. J. Mol. Cell. Cardiol. 2010, 48, 490–496. [Google Scholar] [CrossRef] [Green Version]
- van Hoeven, K.H.; Factor, S.M. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation 1990, 82, 848–855. [Google Scholar] [CrossRef]
- Weber, K.T.; Brilla, C.G.; Janicki, J.S. Myocardial fibrosis: Functional significance and regulatory factors. Cardiovasc. Res. 1993, 27, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.J.; Snorgaard, O.; Faber, J.; Torjesen, P.A.; Hildebrandt, P.; Mehlsen, J.; Hanssen, K.F. Serum levels of advanced glycation end products are associated with left ventricular diastolic function in patients with type 1 diabetes. Diabetes Care 1999, 22, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Norton, G.R.; Candy, G.; Woodiwiss, A.J. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation 1996, 93, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Bauersachs, J. Of mice and men: Models and mechanisms of diabetic cardiomyopathy. Basic Res. Cardiol. 2018, 114, 2. [Google Scholar] [CrossRef] [PubMed]
- Ares-Carrasco, S.; Picatoste, B.; Benito-Martin, A.; Zubiri, I.; Sanz, A.B.; Sanchez-Nino, M.D.; Ortiz, A.; Egido, J.; Tunon, J.; Lorenzo, O. Myocardial fibrosis and apoptosis, but not inflammation, are present in long-term experimental diabetes. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2109–H2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, P.P.; Yang, J.M.; Zhang, M.X.; Zhang, K.; Chen, Y.G.; Zhang, C.; Zhang, Y. Angiotensin-(1-7) treatment mitigates right ventricular fibrosis as a distinctive feature of diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1007–H1019. [Google Scholar] [CrossRef]
- Haider, B.; Yeh, C.K.; Thomas, G.; Oldewurtel, H.A.; Lyons, M.M.; Regan, T.J. Influence of diabetes on the myocardium and coronary arteries of rhesus monkey fed an atherogenic diet. Circ. Res. 1981, 49, 1278–1288. [Google Scholar] [CrossRef]
- Regan, T.J.; Wu, C.F.; Yeh, C.K.; Oldewurtel, H.A.; Haider, B. Myocardial composition and function in diabetes. The effects of chronic insulin use. Circ. Res. 1981, 49, 1268–1277. [Google Scholar] [CrossRef]
- Basu, R.; Oudit, G.Y.; Wang, X.; Zhang, L.; Ussher, J.R.; Lopaschuk, G.D.; Kassiri, Z. Type 1 diabetic cardiomyopathy in the akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2096–H2108. [Google Scholar] [CrossRef]
- Qin, F.; Siwik, D.A.; Luptak, I.; Hou, X.; Wang, L.; Higuchi, A.; Weisbrod, R.M.; Ouchi, N.; Tu, V.H.; Calamaras, T.D.; et al. The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation 2012, 125, 1757–1764,17511 S–S756. [Google Scholar] [CrossRef] [PubMed]
- Sloan, C.; Tuinei, J.; Nemetz, K.; Frandsen, J.; Soto, J.; Wride, N.; Sempokuya, T.; Alegria, L.; Bugger, H.; Abel, E.D. Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted OB/OB mice. Diabetes 2011, 60, 1424–1434. [Google Scholar] [CrossRef]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudina, S.; Bugger, H.; Sena, S.; O’Neill, B.T.; Zaha, V.G.; Ilkun, O.; Wright, J.J.; Mazumder, P.K.; Palfreyman, E.; Tidwell, T.J.; et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009, 119, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by pparalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Benech, J.C.; Benech, N.; Zambrana, A.I.; Rauschert, I.; Bervejillo, V.; Oddone, N.; Damian, J.P. Diabetes increases stiffness of live cardiomyocytes measured by atomic force microscopy nanoindentation. Am. J. Physiol. Cell Physiol. 2014, 307, C910–C919. [Google Scholar] [CrossRef] [PubMed]
- Borbely, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.J.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.; Schalkwijk, C.G.; Bronzwaer, J.G.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef]
- Chen, J.Y.; Tsai, P.J.; Tai, H.C.; Tsai, R.L.; Chang, Y.T.; Wang, M.C.; Chiou, Y.W.; Yeh, M.L.; Tang, M.J.; Lam, C.F.; et al. Increased aortic stiffness and attenuated lysyl oxidase activity in obesity. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 839–846. [Google Scholar] [CrossRef]
- Zhu, Y.; Qiu, H.; Trzeciakowski, J.P.; Sun, Z.; Li, Z.; Hong, Z.; Hill, M.A.; Hunter, W.C.; Vatner, D.E.; Vatner, S.F.; et al. Temporal analysis of vascular smooth muscle cell elasticity and adhesion reveals oscillation waveforms that differ with aging. Aging Cell 2012, 11, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, T.G.; Starodubtseva, M.N.; Yegorenkov, N.I.; Chizhik, S.A.; Zhdanov, R.I. Atomic force microscopy probing of cell elasticity. Micron 2007, 38, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Vinckier, A.; Semenza, G. Measuring elasticity of biological materials by atomic force microscopy. FEBS Lett. 1998, 430, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Suarez, J.; Scott, B.; Dillmann, W.H. Conditional increase in SERCA2a protein is able to reverse contractile dysfunction and abnormal calcium flux in established diabetic cardiomyopathy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1439–R1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trost, S.U.; Belke, D.D.; Bluhm, W.F.; Meyer, M.; Swanson, E.; Dillmann, W.H. Overexpression of the sarcoplasmic reticulum Ca2+-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes 2002, 51, 1166–1171. [Google Scholar] [CrossRef]
- Belke, D.D.; Dillmann, W.H. Altered cardiac calcium handling in diabetes. Curr. Hypertens. Rep. 2004, 6, 424–429. [Google Scholar] [CrossRef]
- Grutzner, A.; Garcia-Manyes, S.; Kotter, S.; Badilla, C.L.; Fernandez, J.M.; Linke, W.A. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys. J. 2009, 97, 825–834. [Google Scholar] [CrossRef]
- Hopf, A.E.; Andresen, C.; Kotter, S.; Isic, M.; Ulrich, K.; Sahin, S.; Bongardt, S.; Roll, W.; Drove, F.; Scheerer, N.; et al. Diabetes-induced cardiomyocyte passive stiffening is caused by impaired insulin-dependent titin modification and can be modulated by neuregulin-1. Circ. Res. 2018, 123, 342–355. [Google Scholar] [CrossRef]
- Khanna, S.; Singh, G.B.; Khullar, M. Nitric oxide synthases and diabetic cardiomyopathy. Nitric Oxide 2014, 43, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Bredt, D.S. nNOS at a glance: Implications for brain and brawn. J. Cell Sci. 2004, 117, 2627–2629. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.; Michel, L.Y.M.; Balligand, J.L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef] [PubMed]
- Pautz, A.; Art, J.; Hahn, S.; Nowag, S.; Voss, C.; Kleinert, H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 2010, 23, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, S.; Cai, L. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. J. Diabetes Investig. 2014, 5, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Cesselli, D.; Jakoniuk, I.; Barlucchi, L.; Beltrami, A.P.; Hintze, T.H.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 2001, 89, 279–286. [Google Scholar] [CrossRef]
- Takano, H.; Zou, Y.; Hasegawa, H.; Akazawa, H.; Nagai, T.; Komuro, I. Oxidative stress-induced signal transduction pathways in cardiac myocytes: Involvement of ROS in heart diseases. Antioxid. Redox Signal. 2003, 5, 789–794. [Google Scholar] [CrossRef]
- Varga, Z.V.; Giricz, Z.; Liaudet, L.; Hasko, G.; Ferdinandy, P.; Pacher, P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta 2015, 1852, 232–242. [Google Scholar] [CrossRef] [Green Version]
- King, M.K.; Coker, M.L.; Goldberg, A.; McElmurray, J.H., 3rd; Gunasinghe, H.R.; Mukherjee, R.; Zile, M.R.; O’Neill, T.P.; Spinale, F.G. Selective matrix metalloproteinase inhibition with developing heart failure: Effects on left ventricular function and structure. Circ. Res. 2003, 92, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Kang, Y.J. Cell death and diabetic cardiomyopathy. Cardiovasc. Toxicol. 2003, 3, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Eckhouse, S.R.; Spinale, F.G. Changes in the myocardial interstitium and contribution to the progression of heart failure. Heart Fail. Clin. 2012, 8, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Diamant, M.; Lamb, H.J.; Smit, J.W.; de Roos, A.; Heine, R.J. Diabetic cardiomyopathy in uncomplicated type 2 diabetes is associated with the metabolic syndrome and systemic inflammation. Diabetologia 2005, 48, 1669–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, O.; Picatoste, B.; Ares-Carrasco, S.; Ramirez, E.; Egido, J.; Tunon, J. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediat. Inflamm. 2011, 2011, 652097. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Rutschow, S.; Jager, S.; Linderer, A.; Anker, S.; Riad, A.; Unger, T.; Schultheiss, H.P.; Pauschinger, M.; Tschope, C. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: The role of angiotensin type 1 receptor antagonism. Diabetes 2007, 56, 641–646. [Google Scholar] [CrossRef]
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Escher, F.; Bucker-Gartner, C.; Spillmann, F.; Noutsias, M.; Riad, A.; Schultheiss, H.P.; et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes 2007, 56, 1834–1841. [Google Scholar] [CrossRef]
- Kibel, A.; Selthofer-Relatic, K.; Drenjancevic, I.; Bacun, T.; Bosnjak, I.; Kibel, D.; Gros, M. Coronary microvascular dysfunction in diabetes mellitus. J. Int. Med. Res. 2017, 45, 1901–1929. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Taqueti, V.R.; Solomon, S.D.; Shah, A.M.; Desai, A.S.; Groarke, J.D.; Osborne, M.T.; Hainer, J.; Bibbo, C.F.; Dorbala, S.; Blankstein, R.; et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur. Heart J. 2018, 39, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H. Energy metabolism of the heart: From basic concepts to clinical applications. Curr. Probl. Cardiol. 1994, 19, 59–113. [Google Scholar] [CrossRef]
- Isfort, M.; Stevens, S.C.; Schaffer, S.; Jong, C.J.; Wold, L.E. Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Glucose transporters in healthy heart and in cardiac disease. Int. J. Cardiol. 2017, 230, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Kraegen, E.W.; Sowden, J.A.; Halstead, M.B.; Clark, P.W.; Rodnick, K.J.; Chisholm, D.J.; James, D.E. Glucose transporters and in vivo glucose uptake in skeletal and cardiac muscle: Fasting, insulin stimulation and immunoisolation studies of GLUT1 and GLUT4. Biochem. J. 1993, 295 Pt 1, 287–293. [Google Scholar] [CrossRef]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.; Abidi, E.; El-Yazbi, A.; Eid, A.; Booz, G.W.; Zouein, F.A. Direct cardiovascular impact of SGLT2 inhibitors: Mechanisms and effects. Heart Fail. Rev. 2018, 23, 419–437. [Google Scholar] [CrossRef]
- Garvey, W.T.; Hardin, D.; Juhaszova, M.; Dominguez, J.H. Effects of diabetes on myocardial glucose transport system in rats: Implications for diabetic cardiomyopathy. Am. J. Physiol 1993, 264, H837–H844. [Google Scholar] [CrossRef]
- Stratmann, B.; Tschoepe, D. The diabetic heart: Sweet, fatty and stressed. Expert Rev. Cardiovasc. Ther. 2011, 9, 1093–1096. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc. Res. 2008, 79, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, J.; Fitscher, B.A.; Ring, A.; Ihl-Vahl, R.; Strasser, R.H.; Stremmel, W. Fatty acid transporters in plasma membranes of cardiomyocytes in patients with dilated cardiomyopathy. Eur. J. Med. Res. 2000, 5, 438–442. [Google Scholar] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Kim, J. Diabetic cardiomyopathy: Where we are and where we are going. Korean J. Intern. Med. 2017, 32, 404–421. [Google Scholar] [CrossRef] [PubMed]
- Chatham, J.C.; Forder, J.R. A 13C-NMR study of glucose oxidation in the intact functioning rat heart following diabetes-induced cardiomyopathy. J. Mol. Cell. Cardiol. 1993, 25, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Wentz, A.E.; d’Avignon, D.A.; Weber, M.L.; Cotter, D.G.; Doherty, J.M.; Kerns, R.; Nagarajan, R.; Reddy, N.; Sambandam, N.; Crawford, P.A. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J. Biol. Chem. 2010, 285, 24447–24456. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, Y.; Wang, Q.; Kralik, P.M.; Epstein, P.N. Causes and characteristics of diabetic cardiomyopathy. Rev. Diabet. Stud. 2006, 3, 108–117. [Google Scholar] [CrossRef]
- Sharma, V.; McNeill, J.H. Diabetic cardiomyopathy: Where are we 40 years later? Can. J. Cardiol. 2006, 22, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Despa, S.; Bers, D.M. Na+ transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, R.; Srodulski, S.; Peng, X.; Margulies, K.B.; Despa, F.; Despa, S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-Glucose cotransport. J. Am. Heart Assoc. 2015, 4, e002183. [Google Scholar] [CrossRef] [PubMed]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Wust, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Martens, P.; Mathieu, C.; Verbrugge, F.H. Promise of SGLT2 inhibitors in heart failure: Diabetes and beyond. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 23. [Google Scholar] [CrossRef]
- Sano, M. Hemodynamic effects of sodium-glucose cotransporter 2 inhibitors. J. Clin. Med. Res. 2017, 9, 457–460. [Google Scholar] [CrossRef]
- Pabel, S.; Wagner, S.; Bollenberg, H.; Bengel, P.; Kovacs, A.; Schach, C.; Tirilomis, P.; Mustroph, J.; Renner, A.; Gummert, J.; et al. Empagliflozin directly improves diastolic function in human heart failure. Eur. J. Heart Fail. 2018, 20, 1690–1700. [Google Scholar] [CrossRef]
- Habibi, J.; Aroor, A.R.; Sowers, J.R.; Jia, G.; Hayden, M.R.; Garro, M.; Barron, B.; Mayoux, E.; Rector, R.S.; Whaley-Connell, A.; et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 2017, 16, 9. [Google Scholar] [CrossRef]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef]
- Labbe, S.M.; Grenier-Larouche, T.; Noll, C.; Phoenix, S.; Guerin, B.; Turcotte, E.E.; Carpentier, A.C. Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 2012, 61, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef]
- Croston, T.L.; Thapa, D.; Holden, A.A.; Tveter, K.J.; Lewis, S.E.; Shepherd, D.L.; Nichols, C.E.; Long, D.M.; Olfert, I.M.; Jagannathan, R.; et al. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H54–H65. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.M.; Mouton, S.; Sebti, Y.; et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV protection in the EMPA-REG outcome trial: A “thrifty substrate” hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 243–251. [Google Scholar] [CrossRef]
- Stanley, W.C.; Meadows, S.R.; Kivilo, K.M.; Roth, B.A.; Lopaschuk, G.D. β-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1626–H1631. [Google Scholar] [CrossRef]
- Suzuki, M.; Takeda, M.; Kito, A.; Fukazawa, M.; Yata, T.; Yamamoto, M.; Nagata, T.; Fukuzawa, T.; Yamane, M.; Honda, K.; et al. Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr. Diabetes 2014, 4, e125. [Google Scholar] [CrossRef]
- Wight, T.N.; Potter-Perigo, S. The extracellular matrix: An active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G950–G955. [Google Scholar] [CrossRef]
- Law, B.; Fowlkes, V.; Goldsmith, J.G.; Carver, W.; Goldsmith, E.C. Diabetes-induced alterations in the extracellular matrix and their impact on myocardial function. Microsc. Microanal. 2012, 18, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Senador, D.; Kanakamedala, K.; Irigoyen, M.C.; Morris, M.; Elased, K.M. Cardiovascular and autonomic phenotype of DB/DB diabetic mice. Exp. Physiol. 2009, 94, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Heickendorff, L.; Ledet, T.; Rasmussen, L.M. Glycosaminoglycans in the human aorta in diabetes mellitus: A study of tunica media from areas with and without atherosclerotic plaque. Diabetologia 1994, 37, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, in press. [Google Scholar] [CrossRef]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Bershadsky, A.; Kozlov, M.; Geiger, B. Adhesion-mediated mechanosensitivity: A time to experiment, and a time to theorize. Curr. Opin. Cell Biol. 2006, 18, 472–481. [Google Scholar] [CrossRef]
- Chen, C.S. Mechanotransduction—A field pulling together? J. Cell Sci. 2008, 121, 3285–3292. [Google Scholar] [CrossRef]
- Traister, A.; Li, M.; Aafaqi, S.; Lu, M.; Arab, S.; Radisic, M.; Gross, G.; Guido, F.; Sherret, J.; Verma, S.; et al. Integrin-linked kinase mediates force transduction in cardiomyocytes by modulating SERCa2a/PLN function. Nat. Commun. 2014, 5, 4533. [Google Scholar] [CrossRef]
- Carter, W.G.; Kaur, P.; Gil, S.G.; Gahr, P.J.; Wayner, E.A. Distinct functions for integrins α3β1 in focal adhesions and α6β4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: Relation to hemidesmosomes. J. Cell Biol. 1990, 111, 3141–3154. [Google Scholar] [CrossRef]
- Teoh, C.M.; Tam, J.K.; Tran, T. Integrin and gpcr crosstalk in the regulation of asm contraction signaling in asthma. J. Allergy 2012, 2012, 341282. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Schwarz, U.S.; Riveline, D.; Goichberg, P.; Tzur, G.; Sabanay, I.; Mahalu, D.; Safran, S.; Bershadsky, A.; Addadi, L.; et al. Force and focal adhesion assembly: A close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 2001, 3, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the linc complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Cadre, B.M.; Qi, M.; Eble, D.M.; Shannon, T.R.; Bers, D.M.; Samarel, A.M. Cyclic stretch down-regulates calcium transporter gene expression in neonatal rat ventricular myocytes. J. Mol. Cell. Cardiol. 1998, 30, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Limbu, S.; Hoang-Trong, T.M.; Prosser, B.L.; Lederer, W.J.; Jafri, M.S. Modeling local x-RoS and calcium signaling in the heart. Biophys. J. 2015, 109, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Hollekim-Strand, S.M.; Bjorgaas, M.R.; Albrektsen, G.; Tjonna, A.E.; Wisloff, U.; Ingul, C.B. High-intensity interval exercise effectively improves cardiac function in patients with type 2 diabetes mellitus and diastolic dysfunction: A randomized controlled trial. J. Am. Coll. Cardiol. 2014, 64, 1758–1760. [Google Scholar] [CrossRef]
- Schmidt, J.F.; Andersen, T.R.; Horton, J.; Brix, J.; Tarnow, L.; Krustrup, P.; Andersen, L.J.; Bangsbo, J.; Hansen, P.R. Soccer training improves cardiac function in men with type 2 diabetes. Med. Sci. Sports Exerc. 2013, 45, 2223–2233. [Google Scholar] [CrossRef]
- Edelmann, F.; Gelbrich, G.; Dungen, H.D.; Frohling, S.; Wachter, R.; Stahrenberg, R.; Binder, L.; Topper, A.; Lashki, D.J.; Schwarz, S.; et al. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: Results of the EX-DHF (exercise training in diastolic heart failure) pilot study. J. Am. Coll. Cardiol. 2011, 58, 1780–1791. [Google Scholar] [CrossRef]
- Chen, B.; Geng, J.; Gao, S.X.; Yue, W.W.; Liu, Q. Eplerenone modulates Interleukin-33/sST2 signaling and IL-1β in left ventricular systolic dysfunction after acute myocardial infarction. J. Interferon Cytokine Res. 2018, 38, 137–144. [Google Scholar] [CrossRef]
- Kim, S.; Yoshiyama, M.; Izumi, Y.; Kawano, H.; Kimoto, M.; Zhan, Y.; Iwao, H. Effects of combination of ace inhibitor and angiotensin receptor blocker on cardiac remodeling, cardiac function, and survival in rat heart failure. Circulation 2001, 103, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, F.; Chen, F.; Jin, Y.; Chen, H.; Liu, D.; Cui, W. Amlodipine and atorvastatin improve ventricular hypertrophy and diastolic function via inhibiting TNF-α, IL-1β and NF-κB inflammatory cytokine networks in elderly spontaneously hypertensive rats. Biomed. Pharmacother. 2016, 83, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Muller-Brunotte, R.; Kahan, T.; Malmqvist, K.; Ring, M.; Edner, M. Tissue velocity echocardiography shows early improvement in diastolic function with irbesartan and atenolol therapy in patients with hypertensive left ventricular hypertrophy. Results form the swedish irbesartan left ventricular hypertrophy investigation vs atenolol (silvhia). Am. J. Hypertens. 2006, 19, 927–936. [Google Scholar]
- Lunder, M.; Janic, M.; Sabovic, M. Prevention of vascular complications in diabetes mellitus patients: Focus on the arterial wall. Curr. Vasc. Pharmacol. 2019, 17, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.; Deo, M.; Cao, A.H.; Hood, S.G.; Huynh, K.; Kiriazis, H.; Du, X.J.; Julius, T.L.; Figtree, G.A.; Dusting, G.J.; et al. Insulin replacement limits progression of diabetic cardiomyopathy in the low-dose streptozotocin-induced diabetic rat. Diabetes Vasc. Dis. Res. 2017, 14, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jodar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef]
- Jojima, T.; Uchida, K.; Akimoto, K.; Tomotsune, T.; Yanagi, K.; Iijima, T.; Suzuki, K.; Kasai, K.; Aso, Y. Liraglutide, a GLP-1 receptor agonist, inhibits vascular smooth muscle cell proliferation by enhancing amp-activated protein kinase and cell cycle regulation, and delays atherosclerosis in apoe deficient mice. Atherosclerosis 2017, 261, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Sposito, A.C.; Berwanger, O.; de Carvalho, L.S.F.; Saraiva, J.F.K. Glp-1ras in type 2 diabetes: Mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc. Diabetol. 2018, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Koska, J.; Sands, M.; Burciu, C.; D’Souza, K.M.; Raravikar, K.; Liu, J.; Truran, S.; Franco, D.A.; Schwartz, E.A.; Schwenke, D.C.; et al. Exenatide protects against glucose- and lipid-induced endothelial dysfunction: Evidence for direct vasodilation effect of glp-1 receptor agonists in humans. Diabetes 2015, 64, 2624–2635. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wu, S.; Guo, S.; Yu, K.; Yang, Z.; Li, L.; Zhang, Y.; Quan, X.; Ji, L.; Zhan, S. Impact of GLP-1 receptor agonists on blood pressure, heart rate and hypertension among patients with type 2 diabetes: A systematic review and network meta-analysis. Diabetes Res. Clin. Pract. 2015, 110, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Sharkovska, Y.; Kalk, P.; Lawrenz, B.; Godes, M.; Hoffmann, L.S.; Wellkisch, K.; Geschka, S.; Relle, K.; Hocher, B.; Stasch, J.P. Nitric oxide-independent stimulation of soluble guanylate cyclase reduces organ damage in experimental low-renin and high-renin models. J. Hypertens. 2010, 28, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Bonderman, D.; Ghio, S.; Felix, S.B.; Ghofrani, H.A.; Michelakis, E.; Mitrovic, V.; Oudiz, R.J.; Boateng, F.; Scalise, A.V.; Roessig, L.; et al. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: A phase iib double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation 2013, 128, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Maggioni, A.P.; Lam, C.S.P.; Pieske-Kraigher, E.; Filippatos, G.; Butler, J.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Scalise, A.V.; et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: Results of the soluble guanylate cyclase stimulator in heart failure patients with preserved ef (socrates-preserved) study. Eur. Heart J. 2017, 38, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Lekakis, J.P.; Nikolaou, M.; Paraskevaidis, I.; Andreadou, I.; Kaplanoglou, T.; Katsimbri, P.; Skarantavos, G.; Soucacos, P.N.; Kremastinos, D.T. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation 2008, 117, 2662–2669. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; Bressi, E.; Marchetti, C.; Carbone, S.; Sonnino, C.; Van Tassell, B.W.; Abbate, A. Interleukin-1β blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J. Cardiovasc. Pharmacol. 2014, 64, 1–6. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Buckley, L.F.; Carbone, S.; Trankle, C.R.; Canada, J.M.; Dixon, D.L.; Abouzaki, N.; Oddi-Erdle, C.; Biondi-Zoccai, G.; Arena, R.; et al. Interleukin-1 blockade in heart failure with preserved ejection fraction: Rationale and design of the diastolic heart failure anakinra response trial 2 (D-HART2). Clin. Cardiol. 2017, 40, 626–632. [Google Scholar] [CrossRef]
- Owens, A.T.; Brozena, S.C.; Jessup, M. New management strategies in heart failure. Circ. Res. 2016, 118, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Gori, M.; D’Elia, E.; Senni, M. Sacubitril/valsartan therapeutic strategy in hfpef: Clinical insights and perspectives. Int. J. Cardiol. 2019, 281, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Malek, V.; Gaikwad, A.B. Telmisartan and thiorphan combination treatment attenuates fibrosis and apoptosis in preventing diabetic cardiomyopathy. Cardiovasc. Res. 2019, 115, 373–384. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolajević Starčević, J.; Janić, M.; Šabovič, M. Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients. Int. J. Mol. Sci. 2019, 20, 1197. https://doi.org/10.3390/ijms20051197
Nikolajević Starčević J, Janić M, Šabovič M. Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients. International Journal of Molecular Sciences. 2019; 20(5):1197. https://doi.org/10.3390/ijms20051197
Chicago/Turabian StyleNikolajević Starčević, Jovana, Miodrag Janić, and Mišo Šabovič. 2019. "Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients" International Journal of Molecular Sciences 20, no. 5: 1197. https://doi.org/10.3390/ijms20051197
APA StyleNikolajević Starčević, J., Janić, M., & Šabovič, M. (2019). Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients. International Journal of Molecular Sciences, 20(5), 1197. https://doi.org/10.3390/ijms20051197