Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process

 , , and
, , and 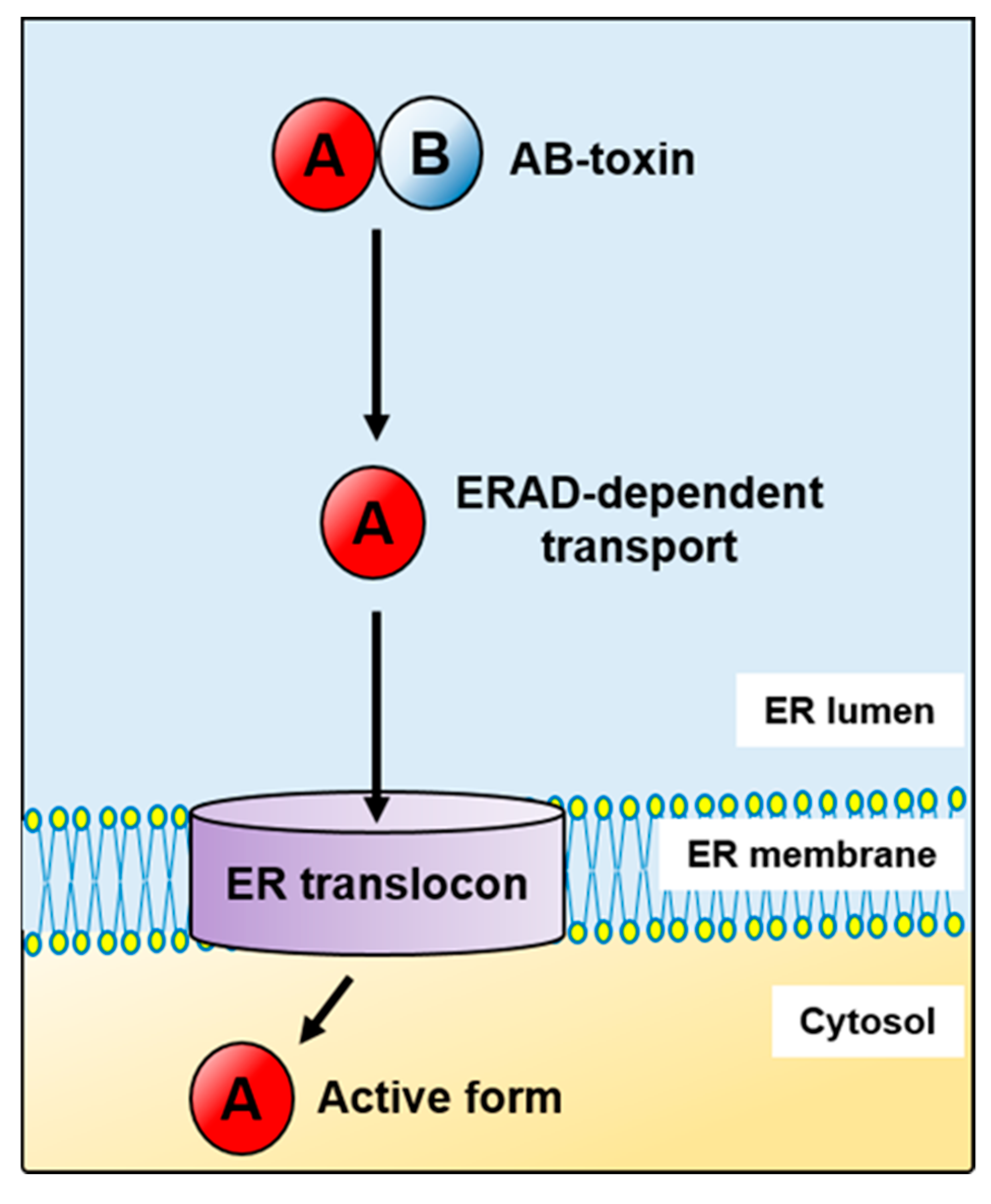{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Toxin Recognition and Disassembly in the ER
2.1. Unfolding and Release of A Fragments from the Holotoxins
2.2. Toxin Interaction with ER Chaperones
2.2.1. Toxin Interaction with Classical ER Chaperones
2.2.2. Toxin Interaction with Carbohydrate-Dependent ER Chaperones
3. Toxin Transport Across the ER Membrane
3.1. Putative ER Retrotranslocons
3.2. Dependence of Toxin A Chain Transport to the Cytosol on the ER Translocon Complexes
4. Toxin Extraction from the ER Membrane, Refolding and Activation in the Cytosol
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Braakman, I.; Hebert, D.N. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Ploegh, H.L. Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol. 2010, 22, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Huyer, G.; Piluek, W.F.; Fansler, Z.; Kreft, S.G.; Hochstrasser, M.; Brodsky, J.L.; Michaelis, S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 2004, 279, 38369–38378. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.; Herzog, V. Endoplasmic reticulum-associated degradation: Exceptions to the rule. Eur. J. Cell Biol. 2004, 83, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Słomińska-Wojewódzka, M.; Sandvig, K. The Role of Lectin-Carbohydrate Interactions in the Regulation of ER-Associated Protein Degradation. Mol. Basel Switz. 2015, 20, 9816–9846. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Omura, S.; Silver, J. Rapid degradation of CD4 in cells expressing human immunodeficiency virus type 1 Env and Vpu is blocked by proteasome inhibitors. J. Gen. Virol. 1997, 78 Pt 3, 619–625. [Google Scholar] [CrossRef]
- Schubert, U.; Antón, L.C.; Bacík, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar]
- Lemberg, M.K.; Freeman, M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007, 17, 1634–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zettl, M.; Adrain, C.; Strisovsky, K.; Lastun, V.; Freeman, M. Rhomboid Family Pseudoproteases Use the ER Quality Control Machinery to Regulate Intercellular Signaling. Cell 2011, 145, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, B.-L.; Sever, N.; DeBose-Boyd, R.A. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 2005, 19, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, S.; Ninagawa, S.; Okada, T.; Koba, H.; Sugimoto, T.; Kamiya, Y.; Kato, K.; Takeda, S.; Mori, K. The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J. Biol. Chem. 2013, 288, 31517–31527. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Rapak, A.; Falnes, P.O.; Olsnes, S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 1997, 94, 3783–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazes, B.; Read, R.J. Accumulating Evidence Suggests That Several AB-Toxins Subvert the Endoplasmic Reticulum-Associated Protein Degradation Pathway to Enter Target Cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Sandvig, K.; Lauvrak, S.U.; van Deurs, B.O. Host cell penetration and trafficking of protein toxins. In Microbial Toxins: Molecular and Cellular Biology; Thomas, P., Ed.; Horizon Press: Norfolk, NJ, USA, 2005; pp. 473–496. [Google Scholar]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Boesze-Battaglia, K.; Alexander, D.; Dlakić, M.; Shenker, B.J. A Journey of Cytolethal Distending Toxins through Cell Membranes. Front. Cell. Infect. Microbiol. 2016, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Kaper, J.B. Enteric bacterial toxins: Mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 1996, 60, 167–215. [Google Scholar] [PubMed]
- Lencer, W.I.; Constable, C.; Moe, S.; Rufo, P.A.; Wolf, A.; Jobling, M.G.; Ruston, S.P.; Madara, J.L.; Holmes, R.K.; Hirst, T.R. Proteolytic activation of cholera toxin and Escherichia coli labile toxin by entry into host epithelial cells. Signal transduction by a protease-resistant toxin variant. J. Biol. Chem. 1997, 272, 15562–15568. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakazawa, T.; Miyata, T.; Kaji, A.; Yokota, T. Evolution and structure of two ADP-ribosylation enterotoxins, Escherichia coli heat-labile toxin and cholera toxin. FEBS Lett. 1984, 169, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.; Vaughan, M. Activation of Adenylate Cyclase by Choleragen. Annu. Rev. Biochem. 1979, 48, 581–600. [Google Scholar] [CrossRef]
- Mudrak, B.; Kuehn, M.J. Heat-Labile Enterotoxin: Beyond GM1 Binding. Toxins 2010, 2, 1445–1470. [Google Scholar] [CrossRef] [Green Version]
- Connell, T.D.; Holmes, R.K. Characterization of hybrid toxins produced in Escherichia coli by assembly of A and B polypeptides from type I and type II heat-labile enterotoxins. Infect. Immun. 1992, 60, 1653–1661. [Google Scholar] [PubMed]
- Paton, J.C.; Paton, A.W. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 1998, 11, 450–479. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Bergan, J.; Dyve, A.-B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon Off. J. Int. Soc. Toxinol. 2010, 56, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Bergan, J.; Dyve Lingelem, A.B.; Simm, R.; Skotland, T.; Sandvig, K. Shiga toxins. Toxicon Off. J. Int. Soc. Toxinol. 2012, 60, 1085–1107. [Google Scholar] [CrossRef]
- Witvliet, M.H.; Burns, D.L.; Brennan, M.J.; Poolman, J.T.; Manclark, C.R. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect. Immun. 1989, 57, 3324–3330. [Google Scholar] [PubMed]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Pihl, A. Different biological properties of the two constituent peptide chains of ricin, a toxic protein inhibiting protein synthesis. Biochemistry 1973, 12, 3121–3126. [Google Scholar] [CrossRef]
- Iglewski, B.H.; Liu, P.V.; Kabat, D. Mechanism of action of Pseudomonas aeruginosa exotoxin Aiadenosine diphosphate-ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144. [Google Scholar]
- Olsnes, S.; Refsnes, K.; Pihl, A. Mechanism of action of the toxic lectins abrin and ricin. Nature 1974, 249, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Hartley, M.R.; Lord, J.M. Cytotoxic ribosome-inactivating lectins from plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar] [CrossRef]
- Eshraghi, A.; Dixon, S.D.; Tamilselvam, B.; Kim, E.J.-K.; Gargi, A.; Kulik, J.C.; Damoiseaux, R.; Blanke, S.R.; Bradley, K.A. Cytolethal Distending Toxins Require Components of the ER-Associated Degradation Pathway for Host Cell Entry. PLoS Pathog. 2014, 10, e1004295. [Google Scholar] [CrossRef]
- Thelestam, M.; Frisan, T. Cytolethal distending toxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 111–133. [Google Scholar]
- McSweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060. [Google Scholar] [CrossRef]
- Nesic, D.; Stebbins, C.E. Mechanisms of Assembly and Cellular Interactions for the Bacterial Genotoxin CDT. PLoS Pathog. 2005, 1, e28. [Google Scholar] [CrossRef] [Green Version]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Sandvig, K.; Garred, O.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef]
- Lencer, W.I.; Constable, C.; Moe, S.; Jobling, M.G.; Webb, H.M.; Ruston, S.; Madara, J.L.; Hirst, T.R.; Holmes, R.K. Targeting of cholera toxin and Escherichia coli heat labile toxin in polarized epithelia: Role of COOH-terminal KDEL. J. Cell Biol. 1995, 131, 951–962. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995, 307 Pt 1, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999, 112 Pt 4, 467–475. [Google Scholar]
- Majoul, I.V.; Bastiaens, P.I.; Söling, H.D. Transport of an external Lys-Asp-Glu-Leu (KDEL) protein from the plasma membrane to the endoplasmic reticulum: Studies with cholera toxin in Vero cells. J. Cell Biol. 1996, 133, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, O.; van Deurs, B. Thapsigargin-induced transport of cholera toxin to the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1996, 93, 12339–12343. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H.; Irish, T.J.; Chen, C.H.; O’Connell, C.B.; Hadley, G.A.; McNamara, U.; Tuskan, R.G.; Lewis, G.K. Intracellular delivery of a cytolytic T-lymphocyte epitope peptide by pertussis toxin to major histocompatibility complex class I without involvement of the cytosolic class I antigen processing pathway. Infect. Immun. 1999, 67, 602–607. [Google Scholar] [PubMed]
- Castro, M.G.; McNamara, U.; Carbonetti, N.H. Expression, activity and cytotoxicity of pertussis toxin S1 subunit in transfected mammalian cells. Cell. Microbiol. 2001, 3, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaut, R.D.; Carbonetti, N.H. Retrograde transport of pertussis toxin in the mammalian cell. Cell. Microbiol. 2008, 10, 1130–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlöw, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Bulleid, N.J. Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2012, 4, a013219. [Google Scholar] [CrossRef]
- Orlandi, P.A. Protein-disulfide Isomerase-mediated Reduction of the A Subunit of Cholera Toxin in a Human Intestinal Cell Line. J. Biol. Chem. 1997, 272, 4591–4599. [Google Scholar]
- Tsai, B.; Rodighiero, C.; Lencer, W.I.; Rapoport, T.A. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 2001, 104, 937–948. [Google Scholar] [CrossRef]
- Forster, M.L.; Sivick, K.; Park, Y.-N.; Arvan, P.; Lencer, W.I.; Tsai, B. Protein disulfide isomerase-like proteins play opposing roles during retrotranslocation. J. Cell Biol. 2006, 173, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Tsai, B.; Rapoport, T.A. Unfolded cholera toxin is transferred to the ER membrane and released from protein disulfide isomerase upon oxidation by Ero1. J. Cell Biol. 2002, 159, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Banerjee, T.; Ray, S.; Tatulian, S.A.; Teter, K. Protein-disulfide Isomerase Displaces the Cholera Toxin A1 Subunit from the Holotoxin without Unfolding the A1 Subunit. J. Biol. Chem. 2011, 286, 22090–22100. [Google Scholar] [CrossRef]
- Taylor, M.; Burress, H.; Banerjee, T.; Ray, S.; Curtis, D.; Tatulian, S.A.; Teter, K. Substrate-induced unfolding of protein disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathog. 2014, 10, e1003925. [Google Scholar] [CrossRef]
- Pande, A.H.; Scaglione, P.; Taylor, M.; Nemec, K.N.; Tuthill, S.; Moe, D.; Holmes, R.K.; Tatulian, S.A.; Teter, K. Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 2007, 374, 1114–1128. [Google Scholar] [CrossRef]
- Massey, S.; Banerjee, T.; Pande, A.H.; Taylor, M.; Tatulian, S.A.; Teter, K. Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J. Mol. Biol. 2009, 393, 1083–1096. [Google Scholar] [CrossRef]
- Cherubin, P.; Guyette, J.; Taylor, M.; O’Donnell, M.; Herndon, L.; Burress, H.; Riad, A.; Tatulian, S.A.; Teter, K. Protein disulfide isomerase does not act as an unfoldase in the disassembly of cholera toxin. Biosci. Rep. 2018, 38, BSR20181320. [Google Scholar] [CrossRef]
- McKee, M.L.; FitzGerald, D.J. Reduction of furin-nicked Pseudomonas exotoxin A: An unfolding story. Biochemistry 1999, 38, 16507–16513. [Google Scholar] [CrossRef]
- Burns, D.L.; Manclark, C.R. Adenine nucleotides promote dissociation of pertussis toxin subunits. J. Biol. Chem. 1986, 261, 4324–4327. [Google Scholar]
- Braakman, I.; Helenius, J.; Helenius, A. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature 1992, 356, 260–262. [Google Scholar] [CrossRef]
- Hazes, B.; Boodhoo, A.; Cockle, S.A.; Read, R.J. Crystal structure of the pertussis toxin-ATP complex: A molecular sensor. J. Mol. Biol. 1996, 258, 661–671. [Google Scholar] [CrossRef]
- Burns, D.L.; Manclark, C.R. Role of cysteine 41 of the A subunit of pertussis toxin. J. Biol. Chem. 1989, 264, 564–568. [Google Scholar]
- Pande, A.H.; Moe, D.; Jamnadas, M.; Tatulian, S.A.; Teter, K. The Pertussis Toxin S1 Subunit Is a Thermally Unstable Protein Susceptible to Degradation by the 20S Proteasome. Biochemistry 2006, 45, 13734–13740. [Google Scholar] [CrossRef]
- Banerjee, T.; Cilenti, L.; Taylor, M.; Showman, A.; Tatulian, S.A.; Teter, K. Thermal Unfolding of the Pertussis Toxin S1 Subunit Facilitates Toxin Translocation to the Cytosol by the Mechanism of Endoplasmic Reticulum-Associated Degradation. Infect. Immun. 2016, 84, 3388–3398. [Google Scholar] [CrossRef] [Green Version]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.H.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef]
- Bellisola, G.; Fracasso, G.; Ippoliti, R.; Menestrina, G.; Rosén, A.; Soldà, S.; Udali, S.; Tomazzolli, R.; Tridente, G.; Colombatti, M. Reductive activation of ricin and ricin A-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharmacol. 2004, 67, 1721–1731. [Google Scholar] [CrossRef]
- Pasetto, M.; Barison, E.; Castagna, M.; Della Cristina, P.; Anselmi, C.; Colombatti, M. Reductive activation of type 2 ribosome-inactivating proteins is promoted by transmembrane thioredoxin-related protein. J. Biol. Chem. 2012, 287, 7367–7373. [Google Scholar] [CrossRef]
- Richardson, P.T.; Westby, M.; Roberts, L.M.; Gould, J.H.; Colman, A.; Lord, J.M. Recombinant proricin binds galactose but does not depurinate 28 S ribosomal RNA. FEBS Lett. 1989, 255, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Mohanraj, D.; Ramakrishnan, S. Cytotoxic effects of ricin without an interchain disulfide bond: Genetic modification and chemical crosslinking studies. Biochim. Biophys. Acta BBA Gen. Subj. 1995, 1243, 399–406. [Google Scholar] [CrossRef]
- Argent, R.H.; Roberts, L.M.; Wales, R.; Robertus, J.D.; Lord, J.M. Introduction of a disulfide bond into ricin A chain decreases the cytotoxicity of the ricin holotoxin. J. Biol. Chem. 1994, 269, 26705–26710. [Google Scholar] [PubMed]
- Argent, R.H.; Parrott, A.M.; Day, P.J.; Roberts, L.M.; Stockley, P.G.; Lord, J.M.; Radford, S.E. Ribosome-mediated folding of partially unfolded ricin A-chain. J. Biol. Chem. 2000, 275, 9263–9269. [Google Scholar] [CrossRef] [PubMed]
- Beddoe, T.; Paton, A.W.; Le Nours, J.; Rossjohn, J.; Paton, J.C. Structure, Biological Functions and Applications of the AB5 Toxins. Trends Biochem. Sci. 2010, 35, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, P.J.; Lingwood, C.A. Membrane cytosolic translocation of verotoxin A1 subunit in target cells. Microbiol. Read. Engl. 2007, 153, 2700–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The Biology of the Cytolethal Distending Toxins. Toxins 2011, 3, 172–190. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Nemec, K.N.; Massey, S.; Tatulian, S.A.; Thelestam, M.; Frisan, T.; Teter, K. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 489–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galligan, J.J.; Petersen, D.R. The human protein disulfide isomerase gene family. Hum. Genom. 2012, 6, 6. [Google Scholar] [CrossRef]
- Irvine, A.G.; Wallis, A.K.; Sanghera, N.; Rowe, M.L.; Ruddock, L.W.; Howard, M.J.; Williamson, R.A.; Blindauer, C.A.; Freedman, R.B. Protein disulfide-isomerase interacts with a substrate protein at all stages along its folding pathway. PLoS ONE 2014, 9, e82511. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Pearse, B.R.; Hebert, D.N. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. N. Y. 2004, 71, 289–297. [Google Scholar]
- Lièvremont, J.P.; Rizzuto, R.; Hendershot, L.; Meldolesi, J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 1997, 272, 30873–30879. [Google Scholar] [CrossRef] [PubMed]
- Kabani, M.; Kelley, S.S.; Morrow, M.W.; Montgomery, D.L.; Sivendran, R.; Rose, M.D.; Gierasch, L.M.; Brodsky, J.L. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol. Biol. Cell 2003, 14, 3437–3448. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. CMLS 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, D.N.; Clerico, E.M.; Gierasch, L.M. Division of Labor: ER-Resident BiP Co-Chaperones Match Substrates to Fates Based on Specific Binding Sequences. Mol. Cell 2016, 63, 721–723. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a Mammalian BiP-associated Protein, Is a Nucleotide Exchange Factor That Regulates the ATPase Activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef] [PubMed]
- Weitzmann, A.; Volkmer, J.; Zimmermann, R. The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett. 2006, 580, 5237–5240. [Google Scholar] [CrossRef]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Breakefield, X.O.; Kamm, C.; Hanson, P.I. TorsinA: Movement at many levels. Neuron 2001, 31, 9–12. [Google Scholar] [CrossRef]
- Winkeler, A.; Gödderz, D.; Herzog, V.; Schmitz, A. BiP-dependent export of cholera toxin from endoplasmic reticulum-derived microsomes. FEBS Lett. 2003, 554, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Dixit, G.; Mikoryak, C.; Hayslett, T.; Bhat, A.; Draper, R.K. Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp. Biol. Med. Maywood NJ 2008, 233, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.; Burress, H.; Taylor, M.; Nemec, K.N.; Ray, S.; Haslam, D.B.; Teter, K. Structural and Functional Interactions between the Cholera Toxin A1 Subunit and ERdj3/HEDJ, a Chaperone of the Endoplasmic Reticulum. Infect. Immun. 2011, 79, 4739–4747. [Google Scholar] [CrossRef]
- Williams, J.M.; Inoue, T.; Banks, L.; Tsai, B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol. Biol. Cell 2013, 24, 785–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.M.; Inoue, T.; Chen, G.; Tsai, B. The nucleotide exchange factors Grp170 and Sil1 induce cholera toxin release from BiP to enable retrotranslocation. Mol. Biol. Cell 2015, 26, 2181–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Easton, D.P.; Chen, X.; MacDonald, I.J.; Wang, X.-Y.; Subjeck, J.R. The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry 2003, 42, 14893–14902. [Google Scholar] [CrossRef]
- Bernardi, K.M.; Forster, M.L.; Lencer, W.I.; Tsai, B. Derlin-1 Facilitates the Retro-Translocation of Cholera Toxin. Mol. Biol. Cell 2008, 19, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.; Bernardi, K.M.; Tsai, B. The Ero1alpha-PDI redox cycle regulates retro-translocation of cholera toxin. Mol. Biol. Cell 2010, 21, 1305–1313. [Google Scholar] [CrossRef]
- Alder, N.N.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, A.; Herrgen, H.; Winkeler, A.; Herzog, V. Cholera Toxin Is Exported from Microsomes by the Sec61p Complex. J. Cell Biol. 2000, 148, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, J.H.; Lizák, B.; Hendershot, L.M. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 2010, 21, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. [Google Scholar] [CrossRef] [PubMed]
- Nery, F.C.; Armata, I.A.; Farley, J.E.; Cho, J.A.; Yaqub, U.; Chen, P.; da Hora, C.C.; Wang, Q.; Tagaya, M.; Klein, C.; et al. TorsinA participates in endoplasmic reticulum-associated degradation. Nat. Commun. 2011, 2, 393. [Google Scholar] [CrossRef]
- Taylor, M.; Navarro-Garcia, F.; Huerta, J.; Burress, H.; Massey, S.; Ireton, K.; Teter, K. Hsp90 Is Required for Transfer of the Cholera Toxin A1 Subunit from the Endoplasmic Reticulum to the Cytosol. J. Biol. Chem. 2010, 285, 31261–31267. [Google Scholar] [CrossRef] [PubMed]
- Falguières, T.; Johannes, L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol. Cell 2006, 98, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, I.; Nakata, M.; Kawabata, S.; Hamada, S. Regulated expression of the Shiga toxin B gene induces apoptosis in mammalian fibroblastic cells. Mol. Microbiol. 1999, 33, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, D.B. Shiga Toxin Is Transported from the Endoplasmic Reticulum following Interaction with the Luminal Chaperone HEDJ/ERdj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, K.; Kamiguchi, K.; Torigoe, T.; Nabeta, C.; Hirohashi, Y.; Asanuma, H.; Tobioka, H.; Koge, N.; Harada, O.; Tamura, Y.; et al. Localization and function in endoplasmic reticulum stress tolerance of ERdj3, a new member of Hsp40 family protein. Cell Stress Chaperones 2004, 9, 253–264. [Google Scholar] [CrossRef]
- Gregers, T.F.; Skånland, S.S.; Wälchli, S.; Bakke, O.; Sandvig, K. BiP negatively affects ricin transport. Toxins 2013, 5, 969–982. [Google Scholar] [CrossRef]
- Chamberlain, K.L.; Marshall, R.S.; Jolliffe, N.A.; Frigerio, L.; Ceriotti, A.; Lord, J.M.; Roberts, L.M. Ricin B chain targeted to the endoplasmic reticulum of tobacco protoplasts is degraded by a CDC48- and vacuole-independent mechanism. J. Biol. Chem. 2008, 283, 33276–33286. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Rapak, A.; Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999, 274, 34443–34449. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Hart, P.J.; Cook, J.P.; Pietroni, P.; Rogon, C.; Höhfeld, J.; Roberts, L.M.; Lord, J.M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 17408–17413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, M.; Srinivas, H.; Kandiah, E.; Gemma, E.; Ellgaard, L.; Oscarson, S.; Helenius, A.; Surolia, A. Interactions of Substrate with Calreticulin, an Endoplasmic Reticulum Chaperone. J. Biol. Chem. 2003, 278, 6194–6200. [Google Scholar] [CrossRef]
- Zielinska, D.F.; Gnad, F.; Wiśniewski, J.R.; Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef]
- Hammond, C.; Braakman, I.; Helenius, A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. USA 1994, 91, 913–917. [Google Scholar] [CrossRef]
- Hebert, D.N.; Foellmer, B.; Helenius, A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 1995, 81, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Ihara, Y.; Cohen-Doyle, M.F.; Saito, Y.; Williams, D.B. Calnexin discriminates between protein conformational states and functions as a molecular chaperone in vitro. Mol. Cell 1999, 4, 331–341. [Google Scholar] [CrossRef]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef]
- Hosokawa, N.; Wada, I.; Hasegawa, K.; Yorihuzi, T.; Tremblay, L.O.; Herscovics, A.; Nagata, K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001, 2, 415–422. [Google Scholar] [CrossRef]
- Mast, S.W.; Diekman, K.; Karaveg, K.; Davis, A.; Sifers, R.N.; Moremen, K.W. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 2005, 15, 421–436. [Google Scholar] [CrossRef]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef]
- Olivari, S.; Molinari, M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS Lett. 2007, 581, 3658–3664. [Google Scholar] [CrossRef] [Green Version]
- Henrissat, B.; Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. [Google Scholar] [CrossRef]
- Cabral, C.M.; Liu, Y.; Sifers, R.N. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem. Sci. 2001, 26, 619–624. [Google Scholar] [CrossRef]
- Hebert, D.N.; Garman, S.C.; Molinari, M. The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005, 15, 364–370. [Google Scholar] [CrossRef]
- Olivari, S.; Cali, T.; Salo, K.E.H.; Paganetti, P.; Ruddock, L.W.; Molinari, M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 2006, 349, 1278–1284. [Google Scholar] [CrossRef]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. EDEM1 accelerates the trimming of alpha1,2-linked mannose on the C branch of N-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Shenkman, M.; Ron, E.; Yehuda, R.; Benyair, R.; Khalaila, I.; Lederkremer, G.Z. Mannosidase activity of EDEM1 and EDEM2 depends on an unfolded state of their glycoprotein substrates. Commun. Biol. 2018, 1, 172. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Ito, S.; Wada, I.; Hosokawa, N. ER-resident protein 46 (ERp46) triggers the mannose-trimming activity of ER degradation-enhancing α-mannosidase-like protein 3 (EDEM3). J. Biol. Chem. 2018, 293, 10663–10674. [Google Scholar] [CrossRef] [PubMed]
- Cormier, J.H.; Tamura, T.; Sunryd, J.C.; Hebert, D.N. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 2009, 34, 627–633. [Google Scholar] [CrossRef]
- Kosmaoglou, M.; Kanuga, N.; Aguilà, M.; Garriga, P.; Cheetham, M.E. A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 2009, 122, 4465–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, E.; Shenkman, M.; Groisman, B.; Izenshtein, Y.; Leitman, J.; Lederkremer, G.Z. Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol. Biol. Cell 2011, 22, 3945–3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.-Y.; Huang, C.-H.; Zhuang, Y.-H.; Christianson, J.C.; Chen, X. EDEM2 and OS-9 are required for ER-associated degradation of non-glycosylated sonic hedgehog. PLoS ONE 2014, 9, e92164. [Google Scholar] [CrossRef] [PubMed]
- Day, P.J.; Owens, S.R.; Wesche, J.; Olsnes, S.; Roberts, L.M.; Lord, J.M. An Interaction between Ricin and Calreticulin That May Have Implications for Toxin Trafficking. J. Biol. Chem. 2001, 276, 7202–7208. [Google Scholar] [CrossRef] [PubMed]
- Slominska-Wojewodzka, M.; Gregers, T.F.; Wälchli, S.; Sandvig, K. EDEM Is Involved in Retrotranslocation of Ricin from the Endoplasmic Reticulum to the Cytosol. Mol. Biol. Cell 2006, 17, 1664–1675. [Google Scholar] [CrossRef] [Green Version]
- Słomińska-Wojewódzka, M.; Pawlik, A.; Sokołowska, I.; Antoniewicz, J.; Węgrzyn, G.; Sandvig, K. The role of EDEM2 compared with EDEM1 in ricin transport from the endoplasmic reticulum to the cytosol. Biochem. J. 2014, 457, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Sominka, H.; Nowakowska-Gołacka, J.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. The role of EDEM3 in ricin cytotoxicity and its transport from the ER to the cytosol. Unpublished. Manuscript in preparation.
- Molinari, M.; Calanca, V.; Galli, C.; Lucca, P.; Paganetti, P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003, 299, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Olivari, S.; Galli, C.; Alanen, H.; Ruddock, L.; Molinari, M. A Novel Stress-induced EDEM Variant Regulating Endoplasmic Reticulum-associated Glycoprotein Degradation. J. Biol. Chem. 2005, 280, 2424–2428. [Google Scholar] [CrossRef]
- Sokołowska, I.; Wälchli, S.; Węgrzyn, G.; Sandvig, K.; Słomińska-Wojewódzka, M. A single point mutation in ricin A-chain increases toxin degradation and inhibits EDEM1-dependent ER retrotranslocation. Biochem. J. 2011, 436, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Sokołowska, I.; Piłka, E.S.; Sandvig, K.; Węgrzyn, G.; Słomińska-Wojewódzka, M. Hydrophobicity of protein determinants influences the recognition of substrates by EDEM1 and EDEM2 in human cells. BMC Cell Biol. 2015, 16, 1. [Google Scholar] [CrossRef]
- Katzin, B.J.; Collins, E.J.; Robertus, J.D. Structure of ricin A-chain at 2.5 A. Proteins 1991, 10, 251–259. [Google Scholar] [CrossRef]
- Yan, Q.; Li, X.-P.; Tumer, N.E. N-glycosylation does not affect the catalytic activity of ricin a chain but stimulates cytotoxicity by promoting its transport out of the endoplasmic reticulum. Traffic 2012, 13, 1508–1521. [Google Scholar] [CrossRef]
- Leto, D.E.; Morgens, D.W.; Zhang, L.; Walczak, C.P.; Elias, J.E.; Bassik, M.C.; Kopito, R.R. Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell 2019, 73, 377–389. [Google Scholar] [CrossRef]
- Simpson, J.C.; Lord, J.M.; Roberts, L.M. Point mutations in the hydrophobic C-terminal region of ricin A chain indicate that Pro250 plays a key role in membrane translocation. Eur. J. Biochem. 1995, 232, 458–463. [Google Scholar] [CrossRef]
- Mayerhofer, P.U.; Cook, J.P.; Wahlman, J.; Pinheiro, T.T.J.; Moore, K.A.H.; Lord, J.M.; Johnson, A.E.; Roberts, L.M. Ricin A Chain Insertion into Endoplasmic Reticulum Membranes Is Triggered by a Temperature Increase to 37 °C. J. Biol. Chem. 2009, 284, 10232–10242. [Google Scholar] [CrossRef]
- Li, X.-P.; Baricevic, M.; Saidasan, H.; Tumer, N.E. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 2007, 75, 417–428. [Google Scholar] [CrossRef]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef]
- Pilon, M.; Schekman, R.; Römisch, K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997, 16, 4540–4548. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Wolf, D.H. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009, 28, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Tretter, T.; Pereira, F.P.; Ulucan, O.; Helms, V.; Allan, S.; Kalies, K.-U.; Römisch, K. ERAD and protein import defects in a sec61 mutant lacking ER-lumenal loop 7. BMC Cell Biol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.-L.; Römisch, K. Proteasome 19S RP Binding to the Sec61 Channel Plays a Key Role in ERAD. PLoS ONE 2015, 10, e0117260. [Google Scholar] [CrossRef] [PubMed]
- Römisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [Green Version]
- Oda, Y.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Nagata, K.; Mori, K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006, 172, 383–393. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Ge, Y.; Zhang, J.; Cao, Y.; Xing, J.; Su, D.; Huang, Y.; Li, M.; Qu, S.; Sun, F.; et al. Derlin-1 promotes ubiquitylation and degradation of the epithelial Na+ channel, ENaC. J. Cell Sci. 2017, 130, 1027–1036. [Google Scholar]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef]
- Stein, A.; Ruggiano, A.; Carvalho, P.; Rapoport, T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014, 158, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef]
- Schoebel, S.; Mi, W.; Stein, A.; Ovchinnikov, S.; Pavlovicz, R.; DiMaio, F.; Baker, D.; Chambers, M.G.; Su, H.; Li, D.; et al. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 2017, 548, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Bays, N.W.; Hampton, R.Y. Cdc48-Ufd1-Npl4: Stuck in the middle with Ub. Curr. Biol. 2002, 12, R366–R371. [Google Scholar] [CrossRef]
- Stolz, A.; Hilt, W.; Buchberger, A.; Wolf, D.H. Cdc48: A power machine in protein degradation. Trends Biochem. Sci. 2011, 36, 515–523. [Google Scholar] [CrossRef]
- Wolf, D.H.; Stolz, A. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 2012, 1823, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Görlich, D.; Rapoport, T.A. Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 1993, 75, 615–630. [Google Scholar] [CrossRef] [Green Version]
- Oliver, J.; Jungnickel, B.; Görlich, D.; Rapoport, T.; High, S. The Sec61 complex is essential for the insertion of proteins into the membrane of the endoplasmic reticulum. FEBS Lett. 1995, 362, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Kalies, K.-U.; Stokes, V.; Hartmann, E. A single Sec61-complex functions as a protein-conducting channel. Biochim. Biophys. Acta 2008, 1783, 2375–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalies, K.U.; Görlich, D.; Rapoport, T.A. Binding of ribosomes to the rought endoplasmic reticulum mediated by the Sec61p-complex. J. Cell Bioll. 1994, 126, 925–934. [Google Scholar] [CrossRef]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Morito, D.; Nagata, K. Pathogenic Hijacking of ER-Associated Degradation: Is ERAD Flexible? Mol. Cell 2015, 59, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.M.; Tyson, J.R.; Reid, P.J.; Stirling, C.J. Distinct domains within yeast Sec61p involved in post-translational translocation and protein dislocation. J. Biol. Chem. 2000, 275, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Elia, F.; Tretter, T.; Romisch, K. The N-terminus of Sec61p plays key roles in ER protein import and ERAD. bioRxiv 2018. [Google Scholar]
- Van den Berg, B.; Clemons, W.M.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein-conducting channel. Nature 2004, 427, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, R.M.; Fernández, I.S.; Scheres, S.H.W.; Hegde, R.S. Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 2014, 157, 1632–1643. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An Update on Sec61 Channel Functions, Mechanisms, and Related Diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef]
- Grotzke, J.E.; Kozik, P.; Morel, J.-D.; Impens, F.; Pietrosemoli, N.; Cresswell, P.; Amigorena, S.; Demangel, C. Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc. Natl. Acad. Sci. USA 2017, 114, E5910–E5919. [Google Scholar] [CrossRef] [Green Version]
- Plemper, R.K.; Bordallo, J.; Deak, P.M.; Taxis, C.; Hitt, R.; Wolf, D.H. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 1999, 112 Pt 22, 4123–4134. [Google Scholar]
- Vashist, S.; Ng, D.T.W. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004, 165, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, R.; Galli, C.; Calanca, V.; Nakajima, T.; Molinari, M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 2010, 188, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Tsai, B.; Arvan, P. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonça, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E.; et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, E582–E591. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Olson, E.M.; Shyng, S.-L. Role of Derlin-1 protein in proteostasis regulation of ATP-sensitive potassium channels. J. Biol. Chem. 2012, 287, 10482–10493. [Google Scholar] [CrossRef]
- Wahlman, J.; DeMartino, G.N.; Skach, W.R.; Bulleid, N.J.; Brodsky, J.L.; Johnson, A.E. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell 2007, 129, 943–955. [Google Scholar] [CrossRef]
- Schwieger, I.; Lautz, K.; Krause, E.; Rosenthal, W.; Wiesner, B.; Hermosilla, R. Derlin-1 and p97/valosin-containing protein mediate the endoplasmic reticulum-associated degradation of human V2 vasopressin receptors. Mol. Pharmacol. 2008, 73, 697–708. [Google Scholar] [CrossRef]
- Chen, S.-F.; Wu, C.-H.; Lee, Y.-M.; Tam, K.; Tsai, Y.-C.; Liou, J.-Y.; Shyue, S.-K. Caveolin-1 interacts with Derlin-1 and promotes ubiquitination and degradation of cyclooxygenase-2 via collaboration with p97 complex. J. Biol. Chem. 2013, 288, 33462–33469. [Google Scholar] [CrossRef]
- Isakov, E.; Stanhill, A. Stalled proteasomes are directly relieved by P97 recruitment. J. Biol. Chem. 2011, 286, 30274–30283. [Google Scholar] [CrossRef]
- Greenblatt, E.J.; Olzmann, J.A.; Kopito, R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant α-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2011, 18, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-H.; Hsiao, H.-T.; Chu, Y.-R.; Ye, Y.; Chen, X. Derlin2 protein facilitates HRD1-mediated retro-translocation of sonic hedgehog at the endoplasmic reticulum. J. Biol. Chem. 2013, 288, 25330–25339. [Google Scholar] [CrossRef]
- Stanley, A.M.; Carvalho, P.; Rapoport, T. Recognition of an ERAD-L substrate analyzed by site-specific in vivo photocrosslinking. FEBS Lett. 2011, 585, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, K.M.; Williams, J.M.; Kikkert, M.; van Voorden, S.; Wiertz, E.J.; Ye, Y.; Tsai, B. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol. Biol. Cell 2010, 21, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Abujarour, R.J.; Dalal, S.; Hanson, P.I.; Draper, R.K. p97 Is in a complex with cholera toxin and influences the transport of cholera toxin and related toxins to the cytoplasm. J. Biol. Chem. 2005, 280, 15865–15871. [Google Scholar] [CrossRef]
- Kothe, M.; Ye, Y.; Wagner, J.S.; De Luca, H.E.; Kern, E.; Rapoport, T.A.; Lencer, W.I. Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J. Biol. Chem. 2005, 280, 28127–28132. [Google Scholar] [CrossRef]
- Saslowsky, D.E.; Cho, J.A.; Chinnapen, H.; Massol, R.H.; Chinnapen, D.J.-F.; Wagner, J.S.; De Luca, H.E.; Kam, W.; Paw, B.H.; Lencer, W.I. Intoxication of zebrafish and mammalian cells by cholera toxin depends on the flotillin/reggie proteins but not Derlin-1 or -2. J. Clin. Invest. 2010, 120, 4399–4409. [Google Scholar] [CrossRef] [Green Version]
- Teter, K. Toxin instability and its role in toxin translocation from the endoplasmic reticulum to the cytosol. Biomolecules 2013, 3, 997–1029. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.C.; Roberts, L.M.; Römisch, K.; Davey, J.; Wolf, D.H.; Lord, J.M. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999, 459, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Moreau, D.; Kumar, P.; Wang, S.C.; Chaumet, A.; Chew, S.Y.; Chevalley, H.; Bard, F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev. Cell 2011, 21, 231–244. [Google Scholar] [CrossRef]
- Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. The role of Sec61 in ricin transport from the ER to the cytosol. Unpublished. Manuscript in preparation.
- Li, S.; Spooner, R.A.; Allen, S.C.H.; Guise, C.P.; Ladds, G.; Schnöder, T.; Schmitt, M.J.; Lord, J.M.; Roberts, L.M. Folding-competent and folding-defective forms of ricin A chain have different fates after retrotranslocation from the endoplasmic reticulum. Mol. Biol. Cell 2010, 21, 2543–2554. [Google Scholar] [CrossRef]
- Redmann, V.; Oresic, K.; Tortorella, L.L.; Cook, J.P.; Lord, M.; Tortorella, D. Dislocation of Ricin Toxin A Chains in Human Cells Utilizes Selective Cellular Factors. J. Biol. Chem. 2011, 286, 21231–21238. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Rogozińska, N.; Sominka, H.; Słomińska-Wojewódzka, M. The role of Derlin proteins in ricin transport from the ER to the cytosol. Unpublished. Manuscript in preparation.
- Dang, H.; Klokk, T.I.; Schaheen, B.; McLaughlin, B.M.; Thomas, A.J.; Durns, T.A.; Bitler, B.G.; Sandvig, K.; Fares, H. Derlin-dependent retrograde transport from endosomes to the Golgi apparatus. Traffic 2011, 12, 1417–1431. [Google Scholar] [CrossRef]
- Koopmann, J.O.; Albring, J.; Hüter, E.; Bulbuc, N.; Spee, P.; Neefjes, J.; Hämmerling, G.J.; Momburg, F. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 2000, 13, 117–127. [Google Scholar] [CrossRef]
- Schäuble, N.; Cavalié, A.; Zimmermann, R.; Jung, M. Interaction of Pseudomonas aeruginosa Exotoxin A with the human Sec61 complex suppresses passive calcium efflux from the endoplasmic reticulum. Channels 2014, 8, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Spooner, R.A.; Hampton, R.Y.; Lord, J.M.; Roberts, L.M. Cytosolic entry of Shiga-like toxin a chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS ONE 2012, 7, e41119. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81. [Google Scholar] [CrossRef]
- Wang, M.; Pickart, C.M. Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 2005, 24, 4324–4333. [Google Scholar] [CrossRef] [Green Version]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Neuber, O.; Jarosch, E.; Volkwein, C.; Walter, J.; Sommer, T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, C.; Buchberger, A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Stach, L.; Freemont, P.S. The AAA+ ATPase p97, a cellular multitool. Biochem. J. 2017, 474, 2953–2976. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.H.; Wang, Y.; Warren, G. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 2002, 21, 5645–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, P.; Schindelin, H. Characterization of an Additional Binding Surface on the p97 N-Terminal Domain Involved in Bipartite Cofactor Interactions. Struct. Lond. Engl. 2016, 24, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pye, V.E.; Beuron, F.; Keetch, C.A.; McKeown, C.; Robinson, C.V.; Meyer, H.H.; Zhang, X.; Freemont, P.S. Structural insights into the p97-Ufd1-Npl4 complex. Proc. Natl. Acad. Sci. USA 2007, 104, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänzelmann, P.; Schindelin, H. Structural Basis of ATP Hydrolysis and Intersubunit Signaling in the AAA+ ATPase p97. Struct. Lond. Engl. 2016, 24, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Shen, Y.; Ballar, P.; Apostolou, A.; Agami, R.; Fang, S. AAA ATPase p97/Valosin-containing Protein Interacts with gp78, a Ubiquitin Ligase for Endoplasmic Reticulum-associated Degradation. J. Biol. Chem. 2004, 279, 45676–45684. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T.A. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadowaki, H.; Satrimafitrah, P.; Takami, Y.; Nishitoh, H. Molecular mechanism of ER stress-induced pre-emptive quality control involving association of the translocon, Derlin-1, and HRD1. Sci. Rep. 2018, 8, 7317. [Google Scholar] [CrossRef] [PubMed]
- Gauss, R.; Sommer, T.; Jarosch, E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006, 25, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Medicherla, B.; Kostova, Z.; Schaefer, A.; Wolf, D.H. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 2004, 5, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsasser, S.; Gali, R.R.; Schwickart, M.; Larsen, C.N.; Leggett, D.S.; Müller, B.; Feng, M.T.; Tübing, F.; Dittmar, G.A.G.; Finley, D. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol. 2002, 4, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, S.; Chandler-Militello, D.; Müller, B.; Hanna, J.; Finley, D. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 2004, 279, 26817–26822. [Google Scholar] [CrossRef] [PubMed]
- Guterman, A.; Glickman, M.H. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J. Biol. Chem. 2004, 279, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Sheaff, R.J.; Singer, J.D.; Swanger, J.; Smitherman, M.; Roberts, J.M.; Clurman, B.E. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol. Cell 2000, 5, 403–410. [Google Scholar] [CrossRef]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta 2014, 1843, 216–221. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Luongo, C.L. Domain-specific bias in arginine/lysine usage by protein toxins. Biochem. Biophys. Res. Commun. 1989, 160, 333–339. [Google Scholar] [CrossRef]
- Worthington, Z.E.V.; Carbonetti, N.H. Evading the proteasome: Absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect. Immun. 2007, 75, 2946–2953. [Google Scholar] [CrossRef] [PubMed]
- Rodighiero, C.; Tsai, B.; Rapoport, T.A.; Lencer, W.I. Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002, 3, 1222–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Herr, R.A.; Chua, W.-J.; Lybarger, L.; Wiertz, E.J.H.J.; Hansen, T.H. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 2007, 177, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Moore, P.; Tsai, B. How viruses and toxins disassemble to enter host cells. Annu. Rev. Microbiol. 2011, 65, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Wernick, N.L.B.; De Luca, H.; Kam, W.R.; Lencer, W.I. N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retrotranslocation from endoplasmic reticulum to cytosol. J. Biol. Chem. 2010, 285, 6145–6152. [Google Scholar] [CrossRef]
- Moore, P.; He, K.; Tsai, B. Establishment of an in vitro transport assay that reveals mechanistic differences in cytosolic events controlling cholera toxin and T-cell receptor α retro-translocation. PLoS ONE 2013, 8, e75801. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.; Lass, A.; Wójcik, C. Ufd1–Npl4 is a negative regulator of cholera toxin retrotranslocation. Biochem. Biophys. Res. Commun. 2007, 355, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, K.M.; Williams, J.M.; Inoue, T.; Schultz, A.; Tsai, B. A deubiquitinase negatively regulates retro-translocation of nonubiquitinated substrates. Mol. Biol. Cell 2013, 24, 3545–3556. [Google Scholar] [CrossRef] [Green Version]
- Burress, H.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Co- and post-translocation roles for HSP90 in cholera Intoxication. J. Biol. Chem. 2014, 289, 33644–33654. [Google Scholar] [CrossRef]
- Ray, S.; Taylor, M.; Banerjee, T.; Tatulian, S.A.; Teter, K. Lipid Rafts Alter the Stability and Activity of the Cholera Toxin A1 Subunit. J. Biol. Chem. 2012, 287, 30395–30405. [Google Scholar] [CrossRef]
- Deeks, E.D.; Cook, J.P.; Day, P.J.; Smith, D.C.; Roberts, L.M.; Lord, J.M. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 2002, 41, 3405–3413. [Google Scholar] [CrossRef]
- Di Cola, A.; Frigerio, L.; Lord, J.M.; Roberts, L.M.; Ceriotti, A. Endoplasmic reticulum-associated degradation of ricin A chain has unique and plant-specific features. Plant Physiol. 2005, 137, 287–296. [Google Scholar] [CrossRef]
- Marshall, R.S.; Jolliffe, N.A.; Ceriotti, A.; Snowden, C.J.; Lord, J.M.; Frigerio, L.; Roberts, L.M. The Role of CDC48 in the Retro-translocation of Non-ubiquitinated Toxin Substrates in Plant Cells. J. Biol. Chem. 2008, 283, 15869–15877. [Google Scholar] [CrossRef] [Green Version]
- Lipson, C.; Alalouf, G.; Bajorek, M.; Rabinovich, E.; Atir-Lande, A.; Glickman, M.; Bar-Nun, S. A proteasomal ATPase contributes to dislocation of endoplasmic reticulum-associated degradation (ERAD) substrates. J. Biol. Chem. 2008, 283, 7166–7175. [Google Scholar] [CrossRef]
- Di Cola, A.; Frigerio, L.; Lord, J.M.; Ceriotti, A.; Roberts, L.M. Ricin A chain without its partner B chain is degraded after retrotranslocation from the endoplasmic reticulum to the cytosol in plant cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14726–14731. [Google Scholar] [CrossRef] [Green Version]
- Pietroni, P.; Vasisht, N.; Cook, J.P.; Roberts, D.M.; Lord, J.M.; Hartmann-Petersen, R.; Roberts, L.M.; Spooner, R.A. The proteasome cap RPT5/Rpt5p subunit prevents aggregation of unfolded ricin A chain. Biochem. J. 2013, 453, 435–445. [Google Scholar] [CrossRef] [Green Version]
- LaPointe, P.; Wei, X.; Gariépy, J. A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J. Biol. Chem. 2005, 280, 23310–23318. [Google Scholar] [CrossRef]
- Menikh, A.; Saleh, M.T.; Gariépy, J.; Boggs, J.M. Orientation in Lipid Bilayers of a Synthetic Peptide Representing the C-Terminus of the A1 Domain of Shiga Toxin. A Polarized ATR-FTIR Study. Biochemistry 1997, 36, 15865–15872. [Google Scholar] [CrossRef]
- Taylor, M.; Banerjee, T.; VanBennekom, N.; Teter, K. Detection of toxin translocation into the host cytosol by surface plasmon resonance. J. Vis. Exp. 2012, e3686. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Jang, J.Y.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Localization of Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin Subunits during Intoxication of Live Cells. Infect. Immun. 2012, 80, 2761–2770. [Google Scholar] [CrossRef] [Green Version]
- McSweeney, L.A.; Dreyfus, L.A. Nuclear localization of the Escherichia coli cytolethal distending toxin CdtB subunit. Cell. Microbiol. 2004, 6, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681. [Google Scholar] [CrossRef]
- Chen, Y.; Bellamy, W.P.; Seabra, M.C.; Field, M.C.; Ali, B.R. ER-associated protein degradation is a common mechanism underpinning numerous monogenic diseases including Robinow syndrome. Hum. Mol. Genet. 2005, 14, 2559–2569. [Google Scholar] [CrossRef] [Green Version]
- Adnan, H.; Zhang, Z.; Park, H.-J.; Tailor, C.; Che, C.; Kamani, M.; Spitalny, G.; Binnington, B.; Lingwood, C. Endoplasmic Reticulum-Targeted Subunit Toxins Provide a New Approach to Rescue Misfolded Mutant Proteins and Revert Cell Models of Genetic Diseases. PLoS ONE 2016, 11, e0166948. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [Green Version]
- Shirley, R.B.; Kaddour-Djebbar, I.; Patel, D.M.; Lakshmikanthan, V.; Lewis, R.W.; Kumar, M.V. Combination of proteasomal inhibitors lactacystin and MG132 induced synergistic apoptosis in prostate cancer cells. Neoplasia 2005, 7, 1104–1111. [Google Scholar] [CrossRef]
- Wu, K.L.; van Wieringen, W.; Vellenga, E.; Zweegman, S.; Lokhorst, H.M.; Sonneveld, P. Analysis of the efficacy and toxicity of bortezomib for treatment of relapsed or refractory multiple myeloma in community practice. Haematologica 2005, 90, 996–997. [Google Scholar]
- Merksamer, P.I.; Papa, F.R. The UPR and cell fate at a glance. J. Cell Sci. 2010, 123, 1003–1006. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Brem, G.J.; Mylonas, I.; Brüning, A. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression. Gynecol. Oncol. 2013, 128, 383–390. [Google Scholar] [CrossRef]
- Słomińska-Wojewódzka, M.; Sandvig, K. Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro. Antibodies 2013, 2, 236–269. [Google Scholar] [CrossRef] [Green Version]
- Antignani, A.; FitzGerald, D. Immunotoxins: The Role of the Toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef]
- Wang, C.-T.; Jetzt, A.E.; Cheng, J.-S.; Cohick, W.S. Inhibition of the Unfolded Protein Response by Ricin A—Chain Enhances Its Cytotoxicity in Mammalian Cells. Toxins 2011, 3, 453–468. [Google Scholar] [CrossRef]
- Parikh, B.A.; Tortora, A.; Li, X.-P.; Tumer, N.E. Ricin Inhibits Activation of the Unfolded Protein Response by Preventing Splicing of the HAC1 mRNA. J. Biol. Chem. 2008, 283, 6145–6153. [Google Scholar] [CrossRef] [Green Version]
- Horrix, C.; Raviv, Z.; Flescher, E.; Voss, C.; Berger, M.R. Plant ribosome-inactivating proteins type II induce the unfolded protein response in human cancer cells. Cell. Mol. Life Sci. 2011, 68, 1269–1281. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef]
- Cho, J.A.; Lee, A.-H.; Platzer, B.; Cross, B.C.S.; Gardner, B.M.; De Luca, H.; Luong, P.; Harding, H.P.; Glimcher, L.H.; Walter, P.; et al. The unfolded protein response element IRE1α senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 2013, 13, 558–569. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowakowska-Gołacka, J.; Sominka, H.; Sowa-Rogozińska, N.; Słomińska-Wojewódzka, M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. Int. J. Mol. Sci. 2019, 20, 1307. https://doi.org/10.3390/ijms20061307
Nowakowska-Gołacka J, Sominka H, Sowa-Rogozińska N, Słomińska-Wojewódzka M. Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. International Journal of Molecular Sciences. 2019; 20(6):1307. https://doi.org/10.3390/ijms20061307
Chicago/Turabian StyleNowakowska-Gołacka, Jowita, Hanna Sominka, Natalia Sowa-Rogozińska, and Monika Słomińska-Wojewódzka. 2019. "Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process" International Journal of Molecular Sciences 20, no. 6: 1307. https://doi.org/10.3390/ijms20061307
APA StyleNowakowska-Gołacka, J., Sominka, H., Sowa-Rogozińska, N., & Słomińska-Wojewódzka, M. (2019). Toxins Utilize the Endoplasmic Reticulum-Associated Protein Degradation Pathway in Their Intoxication Process. International Journal of Molecular Sciences, 20(6), 1307. https://doi.org/10.3390/ijms20061307