Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Intrinsically Disordered Proteins in Some Diseases
2.1. α-Synuclein and Parkinson’s Disease
2.2. Amyloid β-Peptide, Tau Protein, and Alzheimer′s Disease
2.3. Prion Protein in Prion Diseases
2.4. p53, c-Myc, and Cancer
2.5. Amylin and Diabetes
3. Strategies for IDP Modulation
3.1. Regulation of IDP Activity through Structural Changes
3.1.1. Coupled Folding and Binding
3.1.2. Post-Translational Modifications
3.2. Regulation of IDPs Abundance
3.2.1. IDP-Encoding mRNAs
3.2.2. Proteasomal Degradation
3.2.3. Stabilization through “Nanny” Proteins
3.3. Modulation of IDPs by Chaperones and Co-Chaperones
4. Known Drugs Acting on IDPs
5. Status and Challenges in Drug Development for IDPs
6. Conclusion and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| CJD | Creutzfeldt–Jakob Disease |
| CNS | Central Nervous System |
| CS | Conformational Selection |
| GSS | Gerstmann–Sträussler–Scheinker Disease |
| IAPP | Islet Amyloid Polypeptide (Amylin) |
| IDP | Intrinsically Disordered Protein |
| IDR | Intrinsically Disordered Region |
| IF | Induced Fit |
| MT | Microtubules |
| NMR | Nuclear Magnetic Resonance |
| PDB | RCSB Protein Databank |
| PrP | Prion Protein |
| PrPSc | Prion Protein, Alternate Conformation |
| PTM | Post-Translational Modification |
| SLiMs | Short Linear Motifs |
| ssNMR | Solid-State Nuclear Magnetic Resonance |
| TAD | Transactivation Domain (of p53) |
| UD | Ubiquitin-Dependent |
| UI | Ubiquitin-Independent |
References
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, S. Intrinsically disordered proteins: Structural and functional dynamics. Res. Rep. Biol. 2017, 8, 7–16. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding—Misfolding—Nonfolding cross-roads: Which way to go? Cell. Mol. Life Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Daughdrill, G.W.; Pielak, G.J.; Uversky, V.N.; Cortese, M.S.; Dunker, A.K. Natively disordered proteins. In Protein Fold Handbook; Buchner, J., Kiefhaber, T., Eds.; Wiley VCH: Weinheim, Germany, 2005; pp. 275–357. [Google Scholar]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins Struct. Funct. Bioinform. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.W.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 26, S1. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The triple power of D3: Protein intrinsic disorder in degenerative diseases. Front. Biosci. 2014, 19, 181–258. [Google Scholar] [CrossRef]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar] [PubMed]
- Kransnoslobodtsev, A.V.; Shlyakhtenko, L.S.; Ukraintsev, E.; Zaikova, T.O.; Keana, J.F.W.; Lyubchenko, Y.L. Nanomedicine and protein misfolding diseases. Nanomedicine 2005, 1, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the D2 concept. Expert Rev. Proteom. 2010, 7, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Uversky, V.N. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 2011, 3, 1163–1180. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Hyman, B.T.; Wegmann, S. Structural studies on the mechanism of protein aggregation in age related neurodegenerative diseases. Mech. Ageing Dev. 2016, 156, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.; Crowther, R.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.S.; Han, J.Y.; Heo, C.E.; Lee, S.W.; Kim, H.I. Electrostatic and hydrophobic interactions of lipid-associated α-synuclein: The role of a water-limited interfaces in amyloid fibrillation. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [Green Version]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.P.; Yuan, H.X.; Li, X.Q.; Power, J.T.; Blumbergs, P.C.; Jensen, P.H. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000, 166, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Goedert, M.; Spillantini, M.G. The Transcellular Propagation and Intracellular Trafficking of α-Synuclein. Cold Spring Harb. Perspect. Med. 2017, 7, a024380. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, V.R.; Spinelli, K.J.; Weston, L.J.; Luk, K.C.; Woltjer, R.L.; Unni, V.K. Progressive aggregation of alpha-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela, J.A.; Rodrigues, M.; De, S.; Flagmeier, P.; Gandhi, S.; Dobson, C.M.; Klenerman, D.; Lee, S.F. Optical Structural Analysis of Individual α-Synuclein Oligomers. Angew. Chem. Int. Ed. Engl. 2018, 57, 4886–4890. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Aleksis, R.; Oleskovs, F.; Jaudzems, K.; Pahnke, J.; Biverstål, H. Structural studies of amyloid-β peptides: Unlocking the mechanism of aggregation and the associated toxicity. Biochimie 2017, 140, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Rajput, R.; Shrivastava, N.; Somvanshi, P.; Grover, A. Synergistic approaches unraveling regulation and aggregation of intrinsically disordered β-amyloids implicated in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2018, 99, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.; Beher, D.; Cookson, N.; Oakley, A.; Piggott, M.; Morris, C.M.; Jaros, E.; Perry, R.; Ince, P.; Kenny, R.A.; et al. Quantification of Alzheimer pathology in ageing and dementia: Age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 2006, 32, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Sosa, L.J.; Caceres, A.; Dupraz, S.; Oksdath, M.; Quiroga, S.; Lorenzo, A. The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 2017, 143, 11–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldgaber, D.; Lerman, M.I.; McBride, O.W.; Saffiotti, U.; Gajdusek, D.C. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Gusella, J.F.; Watkins, P.C.; Bruns, G.A.; St George-Hyslop, P.; Van Keuren, M.L.; Patterson, D.; Pagan, S.; Kurnit, D.M.; Neve, R.L. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 1987, 235, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Haass, C. Take five—BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Bitan, G.; Vollers, S.S.; Teplow, D.B. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 2003, 12, 34882–34889. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Cheung, T.T.; Cai, X.-D.; Odaka, A.; Otvos, L.; Eckman, C.; Golde, T.E.; Younkin, S.G. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Alexandrescu, A.T. Amyloid accomplices and enforcers. Protein Sci. 2005, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.K.; May, P.C.; Tomaselli, K.J.; Rydel, R.E.; Fuson, K.S.; Brigham, E.F.; Wright, S.; Lieberburg, I.; Becker, G.W.; Brems, D.N. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol. Pharmacol. 1994, 45, 373–379. [Google Scholar] [PubMed]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, C. Aβ42 is More Rigid than Aβ40 at the C Terminus: Implications for Aβ Aggregation and Toxicity. J. Mol. Biol. 2006, 364, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gonnelli, L.; Luchinat, C.; Mao, J.; Nesi, A. A new structural model of Aβ40 fibrils. J. Am. Chem. Soc. 2011, 133, 16013–16022. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Long, F.; Miller, Y.; Xiao, Y.; McElheny, D.; Thurber, K.; Ma, B.; Nussinov, R.; Ishii, Y. Molecular-level examination of Cu2+ binding structure for amyloid fibrils of 40-residue Alzheimer’s β by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 3390–3400. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Yau, W.M.; Qiang, W. Modeling an in-register, parallel “Iowa” Aβ fibril structure using solid-state NMR data from labeled samples with Rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Multhaup, G.; Simms, G.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibriliary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Borna, H.; Assadoulahei, K.; Riazi, G.; Harchegani, A.B.; Shahriary, A. Structure, Function and Interactions of Tau: Particular Focus on Potential Drug Targets for the Treatment of Tauopathies. CNS Neurol. Disord Drug Targets 2018, 17, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Bakota, L.; Ussif, A.; Jeserich, G.; Brandt, R. Systemic and network functions of the microtubule-associated protein tau: Implications for tau-based therapies. Mol. Cell. Neurosci. 2017, 84, 132–141. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4448. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human tau gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Balin, B.J.; Otvos, L., Jr.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, I.; Varghese, M.; Ksiezak-Reding, H.; Dzhun, A.; Wang, J.; Pasinetti, G.M. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J. Biol. Chem. 2012, 287, 20522–20533. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Rank, K.B.; Evans, D.B.; Sharma, S.K. Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem. Biophys. Res. Commun. 2001, 285, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Cehlar, O.; Skrabana, R.; Novak, M. Tau Conformation as a Target for Disease-Modifying Therapy: The Role of Truncation. J. Alzheimers Dis. 2018, 64, S535–S546. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Maria Sciacca, M.F.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Ligabue-Braun, R.; Carlini, C.R. Moonlighting Toxins: Ureases and Beyond. In Plant Toxins; Gopalakrishnakone, P., Carlini, C.R., Ligabue-Braun, R., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 199–219. [Google Scholar]
- Oliveira, J.M.; Henriques, A.G.; Martins, F.; Rebelo, S.; da Cruz e Silva, O.A. Amyloid-beta Modulates Both AbetaPP and Tau Phosphorylation. J. Alzheimers Dis. 2015, 45, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Mckinley, M.P.; Groth, D.F.; Bowman, K.A.; Mock, N.I.; Cochran, S.P.; Masiarz, F.R. Scrapie agent contains a hydrophobic protein. Proc. Natl. Acad. Sci. USA 1981, 78, 6675–6679. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Groth, D.F.; Bolton, D.C.; Kent, S.B.; Hood, L.E. Purification and structural studies of a major scrapie prion protein. Cell 1984, 38, 127–134. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in Disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, A.K.; Jarosz, D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018, 430, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wüthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein PrP (29-231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef] [PubMed]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez-Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-García, F.L.; Zahn, R.; Riek, R.; Wüthrich, K. NMR structure of the bovine prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 8334–8339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Casiraghi, N.; Rossetti, G.; Mohanty, S.; Giachin, G.; Legname, G.; Carloni, P. Role of Prion Disease-Linked Mutations in the Intrinsically Disordered N-Terminal Domain of the Prion Protein. J. Chem. Theory Comput. 2013, 9, 5158–5167. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.-M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.N.A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Chemistry and biology of prions. Biochemistry 1992, 31, 12277–12288. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Gajdusek, D.C. The Human Spongiform Encephalopathies: Kuru, Creutzfeldt-Jakob Disease, and the Gerstmann-Sträussler-Scheinker Syndrome. In Transmissible Spongiform Encephalopathies: Current Topics in Microbiology and Immunology; Chesebro, B.W., Ed.; Springer: Berlin, Germany, 1991; Volume 172, pp. 1–20. [Google Scholar]
- Nathanson, N.; Wilesmith, J.; Griot, C. Bovine spongiform encephalopathy (BSE): Causes and consequences of a common source epidemic. Am. J. Epidemiol. 1997, 145, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Pattison, I.H. The relative susceptibility of sheep, goats and mice to two types of the goat scrapie agent. Res. Vet. Sci. 1966, 7, 207–212. [Google Scholar] [CrossRef]
- Scott, M.; Foster, D.; Mirenda, C.; Serban, D.; Coufal, F.; Wälchli, M.; Torchia, M.; Groth, D.; Carlson, G.; DeArmond, S.J.; et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989, 59, 847–857. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.; Foster, D.; Pan, K.-M.; Groth, D.; Mirenda, C.; Torchia, M.; Yang, S.-L.; Serban, D.; Carlson, G.A.; et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 1990, 63, 673–686. [Google Scholar] [CrossRef]
- Bartz, J.C.; McKenzie, D.I.; Bessen, R.A.; Marsh, R.F.; Aiken, J.M. Transmissible mink encephalopathy species barrier effect between ferret and mink: PrP gene and protein analysis. J. Gen. Virol. 1994, 75, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Khaychuk, V.; Angers, R.C.; Fernández-Borges, N.; Vidal, E.; Meyerett-Reid, C.; Kim, S.; Calvi, C.L.; Bartz, J.C.; Hoover, E.A.; et al. Prion replication without host adaptation during interspecies transmissions. Proc. Natl. Acad. Sci. USA 2017, 114, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Molecular biology and pathogenesis of prion diseases. Trends Biochem. Sci. 1996, 21, 482–487. [Google Scholar] [CrossRef]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, N.; Baldwin, M.A.; Prusiner, S.B.; Teplow, D.B.; Hood, L.; Gibson, B.W.; Burlingame, A.L. Structural Studies of the Scrapie Prion Protein Using Mass Spectrometry and Amino Acid Sequencing. Biochemistry 1993, 32, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.K.; McKinley, M.P.; Bowman, K.A.; Braunfeld, M.B.; Barry, R.A.; Prusiner, S.B. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Saverioni, D.; Notari, S.; Capellari, S.; Poggiolini, I.; Giese, A.; Kretzschmar, H.A.; Parchi, P. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J. Biol. Chem. 2013, 288, 27972–27985. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP Sc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Tzaban, S.; Friedlander, G.; Schonberger, O.; Horonchik, L.; Yedidia, Y.; Shaked, G.; Gabizon, R.; Taraboulos, A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002, 41, 12868–12875. [Google Scholar] [PubMed]
- DeArmond, S.J.; Sánchez, H.; Yehiely, F.; Qiu, Y.; Ninchak-Casey, A.; Daggett, V.; Camerino, A.P.; Cayetano, J.; Rogers, M.; Groth, D.; et al. Selective neuronal targeting in prion disease. Neuron 1997, 19, 1337–1348. [Google Scholar] [CrossRef]
- Colby, D.W.; Zhang, Q.; Wang, S.; Groth, D.; Legname, G.; Riesner, D.; Prusiner, S.B. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20914–20919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, H.; Prusiner, S.B.; Cohen, F.E. Scrapie infectivity is independent of amyloid staining properties of the N-Terminally truncated prion protein. J. Struct. Biol. 2000, 130, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Davé, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Mol, P.R. Oncogenes as Therapeutic Targets in Cancer: A Review. IOSR J. Dent. Med. Sci. 2013, 5, 46–56. [Google Scholar] [CrossRef]
- Dunker, A.K.; Uversky, V.N. Drugs for “protein clouds”: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. Targeting therapies for the p53 protein in cancer treatments. Annu. Rev. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Curtis, C. p53 Mutation Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Müller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003, 332, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [PubMed] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Ernst, J.; Clubb, R.; Omichinski, J.G.; Kennedy, W.M.P.; Sakaguchi, K.; Appella, E.; Gronenborn, A.M. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat. Struct. Mol. Biol. 1995, 2, 321–333. [Google Scholar] [CrossRef]
- Fields, S.; Jang, S.K. Presence of a potent transcription activating sequence in the p53 protein. Science 1990, 249, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Harvey, T.S.; Yin, Y.; Yau, P.; Litchfield, D.; Arrowsmith, C.H. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Mol. Biol. 1994, 1, 877–890. [Google Scholar] [CrossRef]
- Uversky, A.V.; Xue, B.; Peng, Z.; Kurgan, L.; Uversky, V.N. On the intrinsic disorder status of the major players in programmed cell death pathways. F1000Research 2013, 2, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.-C.; Jochemsen, A.G. Mdmx as an essential regulator of p53 activity. Biochem. Biophys. Res. Commun. 2005, 331, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Schon, O.; Friedler, A.; Bycroft, M.; Freund, S.M.V.; Fersht, A.R. Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol. 2002, 323, 491–501. [Google Scholar] [CrossRef]
- Borcherds, W.; Kashtanov, S.; Wu, H.; Daughdrill, G.W. Structural divergence is more extensive than sequence divergence for a family of intrinsically disordered proteins. Proteins Struct. Funct. Bioinform. 2013, 81, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Lee, S.H.; Kim, D.H.; Ahn, M.J.; Kim, J.S.; Woo, J.Y.; Torizawa, T.; Kainosho, M.; Han, K.H. Structural details on mdm2-p53 interaction. J. Biol. Chem. 2005, 280, 38795–38802. [Google Scholar] [CrossRef] [PubMed]
- Popowicz, G.M.; Czarna, A.; Rothweiler, U.; Szwagierczak, A.; Krajewski, M.; Weber, L.; Holak, T.A. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle 2007, 6, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Vise, P.D.; Baral, B.; Latos, A.J.; Daughdrill, G.W. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005, 33, 2061–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlagbauer-Wadl, H.; Griffioen, M.; Van Elsas, A.; Schrier, P.I.; Pustelnik, T.; Eichler, H.; Wolff, K.; Pehamberger, H.; Jansen, B. Influence of Increased c-Myc Expression on the Growth Characteristics of Human Melanoma. J. Investig. Dermatol. 1999, 112, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic interventions of cancers using intrinsically disordered proteins as drug targets: C-myc as model system. Cancer Inform. 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Dalton, S.; Brooks, M.W.; Littlewood, T.D.; Evan, G.I.; Land, H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 1992, 359, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Kretzner, L.; Blackwood, E.M.; Eisenman, R.N.; Weintraub, H. Sequence-specific DNA binding by the c-Myc protein. Science 1990, 250, 1149–1151. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Lee, W.M.; Chen, L.L.; Dang, C.V. Max: Functional domains and interaction with c-Myc. Genes Dev. 1992, 6, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Andresen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.H.; Lundström, P.; et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012, 40, 6353–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, D.Y.L.; Watson, J.D.; Yan, P.S.; Barsyte-Lovejoy, D.; Khosravi, F.; Wong, W.W.-L.; Farnham, P.J.; Huang, T.H.-M.; Penn, L.Z. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 2003, 13, 882–886. [Google Scholar] [CrossRef]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J. Pharmacol. Exp. Ther. 2010, 335, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Raffeiner, P.; Röck, R.; Schraffl, A.; Hartl, M.; Hart, J.R.; Janda, K.D.; Vogt, P.K.; Stefan, E.; Bister, K. In vivo quantification and perturbation of Myc-Max interactions and the impact on oncogenic potential. Oncotarget 2014, 5, 8869–8878. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E. Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: Problems and prospects. Endocr. Rev. 1998, 19, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.J.; Willis, A.C.; Clark, A.; Turner, R.C.; Sim, R.B.; Reid, K.B. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. USA 1987, 84, 8628–8632. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; O’Brien, T.D.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Mosselman, S.; Höppener, J.W.; Zandberg, J.; van Mansfeld, A.D.; Geurts van Kessel, A.H.; Lips, C.J.; Jansz, H.S. Islet amyloid polypeptide: Identification and chromosomal localization of the human gene. FEBS Lett. 1988, 239, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Kapurniotu, A. Amyloidogenicity and cytotoxicity of islet amyloid polypeptide. Biopolymers 2001, 60, 438–459. [Google Scholar] [CrossRef]
- Moore, S.J.; Sonar, K.; Bharadwaj, P.; Deplazes, E.; Mancera, R.L. Characterisation of the Structure and Oligomerisation of Islet Amyloid Polypeptides (IAPP): A Review of Molecular Dynamics Simulation Studies. Molecules 2018, 23, 2142. [Google Scholar] [CrossRef] [PubMed]
- Goldsbury, C.; Goldie, K.; Pellaud, J.; Seelig, J.; Frey, P.; Müller, S.A.; Kistler, J.; Cooper, G.J.; Aebi, U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000, 130, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Kroon, G.J.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 2008, 47, 9900–9910. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Wang, L.; Singh, S.; Ling, Y.L.; Buchanan, L.; Zanni, M.T.; Skinner, J.L.; de Pablo, J.J. Stable and metastable states of human amylin in solution. Biophys. J. 2010, 99, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Bowman, G.R.; Huang, X. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef] [PubMed]
- Höppener, J.W.; Ahrén, B.; Lips, C.J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Höppener, J.W.; Lips, C.J. Role of islet amyloid in type 2 diabetes mellitus. Int. J. Biochem. Cell Biol. 2006, 38, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Qiao, Q.; Qian, Z.; Wei, G. Recent computational studies of membrane interaction and disruption of human islet amyloid polypeptide: Monomers, oligomers and protofibrils. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1826–1839. [Google Scholar] [CrossRef] [PubMed]
- Longhena, F.; Spano, P.; Bellucci, A. Targeting of Disordered Proteins by Small Molecules in Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2018, 245, 85–110. [Google Scholar] [PubMed]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Forman-Kay, J.D. Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J. Making Sense of Intrinsically Disordered Proteins. Biophys. J. 2016, 110, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strome, B.; Hsu, I.S.; Li Cheong Man, M.; Zarin, T.; Nguyen Ba, A.; Moses, A.M. Short linear motifs in intrinsically disordered regions modulate HOG signaling capacity. BMC Syst. Biol. 2018, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011, 21, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional analysis of human hub proteins and their interactors involved in the intrinsic disorder-enriched interactions. Int. J. Mol. Sci. 2017, 18, 2761. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Kovacs, D. Intrinsically disordered chaperones in plants and animals. Biochem. Cell Biol. 2010, 88, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, M.A.; Ellenberger, T.; Wobbe, C.R.; Lee, J.P.; Harrison, S.C.; Struhl, K. Folding transition in the DMA-binding domain of GCN4 on specific binding to DNA. Nature 1990, 347, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.; Carr, P.A.; Cavanagh, J.; Palmer, A.G. Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: Implications for the entropy of association with DNA. J. Mol. Biol. 1999, 285, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Shammas, S.L.; Crabtree, M.D.; Dahal, L.; Wicky, B.I.M.; Clarke, J. Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem. 2016, 291, 6689–6695. [Google Scholar] [CrossRef] [PubMed]
- Hammes, G.G.; Chang, Y.-C.; Oas, T.G. Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 13737–13741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeForte, S.; Uversky, V.N. Order, disorder, and everything in between. Molecules 2016, 21, 1090. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, M.; Biernat, J.; Bibow, S.; Ozenne, V.; Jensen, M.R.; Kadavath, H.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Phosphorylation of human tau protein by microtubule affinity-regulating kinase 2. Biochemistry 2013, 52, 9068–9079. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Ferreira, A.M.; Otieno, S.; Xiao, L.; Bashford, D.; Kriwacki, R.W. Incomplete folding upon binding mediates Cdk4/cyclin D complex activation by tyrosine phosphorylation of inhibitor p27 protein. J. Biol. Chem. 2011, 286, 30142–30151. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; He, Y.; Yang, F.; Mooney, S.M.; Getzenberg, R.H.; Orban, J.; Kulkarni, P. The cancer/testis antigen prostate-associated gene 4 (PAGE4) is a highly intrinsically disordered protein. J. Biol. Chem. 2011, 286, 13985–13994. [Google Scholar] [CrossRef] [PubMed]
- Coskuner-Weber, O.; Uversky, V.N. Insights into the molecular mechanisms of Alzheimer’s and Parkinson’s diseases with molecular simulations: Understanding the roles of artificial and pathological missense mutations in intrinsically disordered proteins related to pathology. Int. J. Mol. Sci. 2018, 19, 336. [Google Scholar] [CrossRef] [PubMed]
- Edwards, Y.J.K.; Lobley, A.E.; Pentony, M.M.; Jones, D.T. Insights into the regulation of intrinsically disordered proteins in the human proteome by analyzing sequence and gene expression data. Genome Biol. 2009, 10, R50. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.G.; Jin, A.J.; Ma, K.; Gutierrez-Cruz, G.; Tsai, W.L.; Wang, K. Titin PEVK segment: Charge-driven elasticity of the open and flexible polyampholyte. J. Muscle Res. Cell Motil. 2005, 26, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Boothby, T.C.; Tapia, H.; Brozena, A.H.; Piszkiewicz, S.; Smith, A.E.; Giovannini, I.; Rebecchi, L.; Pielak, G.J.; Koshland, D.; Goldstein, B. Tardigrades Use Intrinsically Disordered Proteins to Survive Desiccation. Mol. Cell 2017, 65, 975.e5–984.e5. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Olivas, W.M. Conditional regulation of Puf1p, Puf4p, and Puf5p activity alters YHB1 mRNA stability for a rapid response to toxic nitric oxide stress in yeast. Mol. Biol. Cell 2015, 26, 1015–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Ogé, L.; Perez-Garcia, M.D.; Hamama, L.; Sakr, S. The PUF protein family: Overview on PUF RNA targets, biological functions, and post transcriptional regulation. Int. J. Mol. Sci. 2018, 19, 410. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. The nanny model for IDPs. Nat. Chem. Biol. 2009, 5, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Inobe, T.; Matouschek, A. Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 2014, 24, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagai, T.; Azia, A.; Tóth-Petróczy, Á.; Levy, Y. Intrinsic disorder in ubiquitination substrates. J. Mol. Biol. 2011, 412, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.; Baumeister, W. Conformational constraints in protein degradation by the 20S proteasome. Nat. Struct. Biol. 1995, 2, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Binolfi, A.; Frembgen-Kesner, T.; Hingorani, K.; Sarkar, M.; Kyne, C.; Li, C.; Crowley, P.B.; Gierasch, L.; Pielak, G.J.; et al. Physicochemical Properties of Cells and Their Effects on Intrinsically Disordered Proteins (IDPs). Chem. Rev. 2014, 114, 6661–6714. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Tan, M.S.; Lu, R.C.; Yu, J.T.; Tan, L. Heat shock proteins at the crossroads between cancer and Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 239164. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. 2009, 17, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Chang, X.; Ma, D. Hsp90 maintains the stability and function of the tau phosphorylating kinase GSK3β. Int. J. Mol. Sci 2007, 8, 51–60. [Google Scholar] [CrossRef]
- Shelton, L.B.; Baker, J.D.; Zheng, D.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J.; et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 9707–9712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Young, Z.T.; Rauch, J.N.; Assimon, V.A.; Jinwal, U.K.; Ahn, M.; Li, X.; Dunyak, B.M.; Ahmad, A.; Carlson, G.A.; Srinivasan, S.R.; et al. Stabilizing the Hsp70-Tau complex promotes turnover in models of Tauopathy. Cell Chem. Biol. 2016, 23, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, B.; Chapple, J.P.; van der Spuy, J.; Höhfeld, J.; Cheetham, M.E. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 2005, 15, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Schmid, F.X.; Fischer, G. Catalysis of protein folding by prolyl isomerase. Nature 1987, 329, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [PubMed]
- Torbeev, V.Y.; Hilvert, D. Both the cis-trans equilibrium and isomerization dynamics of a single proline amide modulate β2-microglobulin amyloid assembly. Proc. Natl. Acad. Sci. USA 2013, 110, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.D.; Shelton, L.B.; Zheng, D.; Favretto, F.; Nordhues, B.A.; Darling, A.; Sullivan, L.E.; Sun, Z.; Solanki, P.K.; Martin, M.D.; et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017, 15, e2001336. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010, 17, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.J.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Cao, Z.X.; Zhao, L.L.; Li, S.Q. Novel strategies for drug discovery based on intrinsically disordered proteins (IDPs). Int. J. Mol. Sci. 2011, 12, 3205–3219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, H.; Liu, Z. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. 2015, 24, 688–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Huang, Y. Advantages of proteins being disordered. Protein Sci. 2014, 23, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; De Simone, A.; Schenk, D.; Toth, G.; Dobson, C.M.; Vendruscolo, M. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 2013, 139, 035101. [Google Scholar] [CrossRef] [PubMed]
- Fokkens, M.; Schrader, T.; Klärner, F.G. A molecular tweezer for lysine and arginine. J. Am. Chem. Soc. 2005, 127, 14415–14421. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lopes, D.H.; Du, Z.; Pang, E.; Shanmugam, A.; Lomakin, A.; Talbiersky, P.; Tennstaedt, A.; McDaniel, K.; Bakshi, R.; et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 2011, 133, 16958–16969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hare, E.; Scopes, D.I.; Kim, E.M.; Palmer, P.; Spanswick, D.; McMahon, B.; Amijee, H.; Nerou, E.; Treherne, J.M.; Jeggo, R. Novel 5-aryloxypyrimidine SEN1576 as a candidate for the treatment of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2014, 17, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Prabhudesai, S.; Sinha, S.; Attar, A.; Kotagiri, A.; Fitzmaurice, A.G.; Lakshmanan, R.; Ivanova, M.I.; Loo, J.A.; Klärner, F.G.; Schrader, T.; et al. A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 2012, 9, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Sievers, S.A.; Karanicolas, J.; Chang, H.W.; Zhao, A.; Jiang, L.; Zirafi, O.; Stevens, J.T.; Münch, J.; Baker, D.; Eisenberg, D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Frenkel-Pinter, M.; Tal, S.; Scherzer-Attali, R.; Abu-Hussien, M.; Alyagor, I.; Eisenbaum, T.; Gazit, E.; Segal, D. Naphthoquinone-Tryptophan Hybrid Inhibits Aggregation of the Tau-Derived Peptide PHF6 and Reduces Neurotoxicity. J. Alzheimers Dis. 2016, 51, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Njomen, E.; Sjögren, B.; Dexheimer, T.S.; Tepe, J.J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017, 12, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Ni, M.; Li, J.K.; Zhang, Y.P.; Ouyang, Q.; Tang, C. Decision making of the p53 network: Death by integration. J. Theor. Biol. 2011, 271, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Q.; Liu, Z.R. Anchoring intrinsically disordered proteins to multiple targets: Lessons from N terminus of the p53 protein. Int. J. Mol. Sci. 2011, 12, 1410–1430. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.L.; Vu, B.T.; Qing, W.G.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress? Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Stover, J.S.; Whitby, L.R.; Vogt, P.K.; Boger, D.L. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg. Med. Chem. Lett. 2009, 19, 6038–6041. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.Y.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef] [PubMed]
- Zirath, H.; Frenzel, A.; Oliynyk, G.; Segerstrom, L.; Westermark, U.K.; Larsson, K.; Persson, M.M.; Hultenby, K.; Lehtio, J.; Einvik, C.; et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10258–10263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Niu, X.; Jin, F.; Liu, Z.; Jin, C.; Lai, L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016, 6, 22298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkizan, H.V.; Kong, Y.L.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.S.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Youbi, S.E.; Hong, S.P.; Kallakury, B.; Monroe, P.; Erkizan, H.V.; Barber-Rotenberg, J.S.; Houghton, P.; Uren, A.; Toretsky, J.A. Pharmacokinetic modeling optimizes inhibition of the ’undruggable’ EWS-FLI1 transcription factor in Ewing Sarcoma. Oncotarget 2014, 5, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, H.; Buday, L.; Tompa, P. Intrinsic structural disorder confers cellular viability on oncogenic fusion proteins. PLoS Comput. Biol. 2009, 5, e1000552. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Q.; Liu, Z.R. Do intrinsically disordered proteins possess high specificity in protein-protein interactions? Chem.-Eur. J. 2013, 19, 4462–4467. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.S.; Nesbit, J.B.; Marrero, L.; Erfurth, F.; LaRussa, V.F.; Hemenway, C.S. The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia 2004, 18, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Palermo, C.M.; Bennett, C.A.; Winters, A.C.; Hemenway, C.S. The AF4-mimetic peptide, PFWT, induces necrotic cell death in MV4–11 leukemia cells. Leuk. Res. 2008, 32, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Watson, V.G.; Drake, K.M.; Peng, Y.; Napper, A.D. Development of a high-throughput screening-compatible assay for the discovery of inhibitors of the AF4-AF9 interaction using AlphaScreen technology. Assay Drug Dev. Technol. 2013, 11, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002, 1, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Männistö, B.T.; Maguire-Zeiss, K.A.; Myöhänen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Svarcbahs, R.; Julku, U.H.; Myöhänen, T.T. Inhibition of Prolyl Oligopeptidase Restores Spontaneous Motor Behavior in the α-Synuclein Virus Vector-Based Parkinson’s Disease Mouse Model by Decreasing α-Synuclein Oligomeric Species in Mouse Brain. J. Neurosci. 2016, 36, 12485–12497. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Bötzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Hefti, F.; Tsubery, H.; Lulu, M.; Proschitsky, M.; Fisher, R. Conformation as the Therapeutic Target for Neurodegenerative Diseases. Curr. Alzheimer Res. 2017, 14, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; Schroeter, S.; Carroll, J.C.; Cullen, V.; Asp, E.; Proschitsky, M.; Chung, C.H.; Gilead, S.; Nadeem, M.; Dodiya, H.B.; et al. NPT088 reduces both amyloid-β and tau pathologies in transgenic mice. Alzheimers Dement. 2016, 2, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, B.; Sierks, M.R. Intracellular targeting and clearance of oligomeric alpha-synuclein alleviates toxicity in mammalian cells. Neurosci. Lett. 2009, 459, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, M.A.; Messer, A.; Kordower, J.H. Can intrabodies serve as neuroprotective therapies for Parkinson’s disease? Beginning thoughts. J. Parkinsons Dis. 2013, 3, 581–591. [Google Scholar] [PubMed]
- Emadi, S.; Liu, R.; Yuan, B.; Schulz, P.; McAllister, C.; Lyubchenko, Y.; Messer, A.; Sierks, M.R. Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry 2004, 43, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.C.; Joshi, S.N.; Genst, E.; Baghel, A.S.; Dobson, C.M.; Messer, A. Bifunctional Anti-Non-Amyloid Component α-Synuclein Nanobodies Are Protective In Situ. PLoS ONE 2016, 11, e0165964. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. NPJ Parkinsons Dis. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.; Merchant, K.; Bezard, E.; et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-α-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, D.M.; Kalia, S.K.; Kalia, L.V. Emerging disease-modifying strategies targeting alpha-synuclein for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2018, 175, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Csizmadia, G.; Solti, K.; Keresztes, A.; Zhu, M.; Pickhardt, M.; Mandelkow, E.; Toth, G. Structural Basis of Small Molecule Targetability of Monomeric Tau Protein. ACS Chem. Neurosci. 2018, 9, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Pickhardt, M.; Neumann, T.; Schwizer, D.; Callaway, K.; Vendruscolo, M.; Schenk, D.; St George-Hyslop, P.; Mandelkow, E.M.; Dobson, C.M.; McConlogue, L.; et al. Identification of Small Molecule Inhibitors of Tau Aggregation by Targeting Monomeric Tau As a Potential Therapeutic Approach for Tauopathies. Curr. Alzheimer Res. 2015, 12, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Baggett, D.W.; Nath, A. The Rational Discovery of a Tau Aggregation Inhibitor. Biochemistry 2018, 57, 6099–6107. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, N.; Pikman, R.; Giladi, E.; Gozes, I. Protection against tauopathy by the drug candidates NAP (davunetide) and D-SAL: Biochemical, cellular and behavioral aspects. Curr. Pharm. Des. 2011, 17, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Ivashko-Pachima, Y.; Gozes, I. NAP protects against Tau hyperphosphorylation through GSK3. Curr. Pharm. Des. 2018, 24, 3868–3877. [Google Scholar] [CrossRef] [PubMed]
- Dammers, C.; Yolcu, D.; Kukuk, L.; Willbold, D.; Pickhardt, M.; Mandelkow, E.; Horn, A.H.; Sticht, H.; Malhis, M.N.; Will, N.; et al. Selection and Characterization of Tau Binding -Enantiomeric Peptides with Potential for Therapy of Alzheimer Disease. PLoS ONE 2016, 11, e0167432. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, E.; Choi, W.H.; Lee, J.; Lee, J.H.; Lee, H.; Kim, D.E.; Suh, Y.H.; Lee, M.J. Inhibitory RNA Aptamers of Tau Oligomerization and Their Neuroprotective Roles against Proteotoxic Stress. Mol. Pharm. 2016, 13, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, S.; Asadollahi, K.; Riazi, G.; Ahmadian, S.; Saboury, A.A. Vitamin B12 Inhibits Tau Fibrillization via Binding to Cysteine Residues of Tau. ACS Chem. Neurosci. 2017, 8, 2676–2682. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, J.; Soeda, Y.; Ida, T.; Sumioka, A.; Yoshikawa, M.; Matsushita, K.; Akaike, T.; Takashima, A. Modification of Tau by 8-Nitroguanosine 3,5-Cyclic Monophosphate (8-Nitro-cGMP): Effects of nitric oxide-linked chemical modification on tau aggregation. J. Biol. Chem. 2016, 291, 22714–22720. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Lee, S.; Huang, X.; Liu, S.; Inayathullah, M.; Kim, K.-M.; Tang, H.; Ashford, J.W.; Rajadas, J. Attenuation of synaptic toxicity and MARK4/PAR1-mediated Tau phosphorylation by methylene blue for Alzheimer’s disease treatment. Sci. Rep. 2016, 6, 34784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, R.C.; Lew, J.; Graves, D.J. Interaction of Cinnamaldehyde and Epicatechin with Tau: Implications of Beneficial Effects in Modulating Alzheimer’s Disease Pathogenesis. J. Alzheimers Dis. 2013, 36, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martinez-Gonzalez, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3 beta and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Avila, C.; Sanchez-Martinez, M. Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study. Mar. Drugs 2018, 16, 386. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.E. Beta-secretase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Mead, E.; Kestoras, D.; Gibson, Y.; Hamilton, L.; Goodson, R.; Jones, S.; Eversden, S.; Davies, P.; O’Neill, M.; Hutton, M.; et al. Halting of Caspase Activity Protects Tau from MC1-Conformational Change and Aggregation. J. Alzheimers Dis. 2016, 54, 1521–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.V.; McBrayer, M.K.; Campbell, J.; Kumar, A.; Hashim, A.; Sershen, H.; Stavrides, P.H.; Ohno, M.; Hutton, M.; Nixon, R.A. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J. Neurosci. 2014, 34, 9222–9234. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Novak, P.; Novak, M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res. Ther. 2014, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Kontsekova, E.; Zilka, N.; Novak, M. Ten Years of Tau-Targeted Immunotherapy: The Path Walked and the Roads Ahead. Front. Neurosci. 2018, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Shahpasand, K.; Sepehri Shamloo, A.; Nabavi, S.M.; Lu, K.P.; Zhou, X.Z. Tau immunotherapy: Hopes and hindrances. Hum. Vaccin Immunother. 2018, 14, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Cehlar, O.; Skrabana, R.; Kovac, A.; Kovacech, B.; Novak, M. Crystallization and preliminary X-ray diffraction analysis of tau protein microtubule-binding motifs in complex with Tau5 and DC25 antibody Fab fragments. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Tortelli, R.; Galizia, I.; Prete, C.; Daniele, A.; et al. Tau-based therapeutics for Alzheimer’s disease: Active and passive immunotherapy. Immunotherapy 2016, 8, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Zilka, N.; Zilkova, M.; Kovacech, B.; Skrabana, R.; Ondrus, M.; Fialova, L.; Kontsekova, E.; Otto, M.; Novak, M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimers Dis. 2019, 6, 63–69. [Google Scholar] [PubMed]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Zilka, N.; Kovacech, B.; Skrabana, R.; Vince-Kazmerova, Z.; Katina, S.; Fialova, L.; Prcina, M.; et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017, 16, 123–134. [Google Scholar] [CrossRef]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Kovacech, B.; Smolek, T.; Katina, S.; Fialova, L.; Prcina, M.; Parrak, V.; Dal-Bianco, P.; et al. FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; Lopez-Deber, M.; Reis, P.; Hickman, D.; Adolfsson, O.; Chuard, N.; Ndao, D.; et al. Efficacy and Safety of A Liposome-Based Vaccine against Protein Tau, Assessed in Tau.P301L Mice That Model Tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Jiang, H.; Mahan, T.; Maloney, S.; Wozniak, D.; Diamond, M.; Holtzmanm, D. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Trans. Neurol. 2016, 2, 278–288. [Google Scholar] [CrossRef] [PubMed]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J. Prev. Alzheimers Dis. 2017, 4, 236–241. [Google Scholar] [PubMed]
- Rosenberg, R.N.; Fu, M.; Lambracht-Washington, D. Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimers Res. Ther. 2018, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.L.; Tung, Y.C.; Liu, F.; Gong, C.X.; Iqbal, K. Tau passive immunization inhibits not only tau but also Aβ pathology. Alzheimers Res. Ther. 2017, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hol, W.G.J. Protein Crystallography and Computer Graphics—Toward Rational Drug Design. Angew. Chem. 1986, 25, 767–778. [Google Scholar] [CrossRef]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Vendruscolo, M. Druggability of Intrinsically Disordered Proteins. Adv. Exp. Med. Biol. 2015, 870, 383–400. [Google Scholar] [PubMed]
- Marasco, D.; Scognamiglio, P.L. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int. J. Mol. Sci. 2015, 16, 7394–7412. [Google Scholar] [CrossRef] [PubMed]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Ghaleh, N.; Blackledge, M.; Zweckstetter, M. Intrinsically disordered proteins: From sequence and conformational properties toward drug discovery. ChemBioChem 2012, 13, 930–950. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J. Biol. Chem. 2016, 291, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Yu, C.; Lai, L.; Liu, Z. Ligand clouds around protein clouds: A scenario of ligand binding with intrinsically disordered proteins. PLoS Comput. Biol. 2013, 9, e1003249. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.; Thiel, P.; Briels, J.; Ottmann, C. Stabilization of Protein-Protein Interactions in chemical biology and drug discovery. Prog. Biophys. Mol. Biol. 2015, 119, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Drews, J.; Ryser, S. The role of innovation in drug development. Nat. Biotechnol. 1997, 15, 1318–1319. [Google Scholar] [CrossRef] [PubMed]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, Y.; Wang, X.; Li, J.; Liu, W.; Rong, L.; Bao, J. An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014. Int. J. Mol. Sci. 2015, 16, 23446–23562. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.T.; Aprile, F.A.; Vendruscolo, M. Methods of probing the interactions between small molecules and disordered proteins. Cell. Mol. Life Sci. 2017, 74, 3225–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J.; Cragnell, C.; Skepö, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput. 2015, 11, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Dogan, J.; Gianni, S.; Jemth, P. The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, N.C.; Kikuchi, M. Structural flexibility of intrinsically disordered proteins induces stepwise target recognition. J. Chem. Phys. 2013, 139, 225103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammak, S.; Zinzalla, G. Targeting protein-protein interactions (PPIs) of transcription factors: Challenges of intrinsically disordered proteins (IDPs) and regions (IDRs). Prog. Biophys. Mol. Biol. 2015, 119, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Hausrath, A.C.; Kingston, R.L. Conditionally disordered proteins: Bringing the environment back into the fold. Cell. Mol. Life Sci. 2017, 74, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, G.; Åberg, E.; Camilloni, C.; Sundell, G.N.; Andersson, E.; Dogan, J.; Chi, C.N.; Vendruscolo, M.; Jemth, P. Emergence and evolution of an interaction between intrinsically disordered proteins. eLife 2017, 6, e16059. [Google Scholar] [CrossRef] [PubMed]
- De Cássia Ruy, P.; Torrieri, R.; Toledo, J.S.; de Souza Alves, V.; Cruz, A.K.; Ruiz, J.C. Intrinsically disordered proteins (IDPs) in trypanosomatids. BMC Genom. 2014, 15, 1100. [Google Scholar]
- Longhi, S. Structural disorder within paramyxoviral nucleoproteins. FEBS Lett. 2015, 589, 2649–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.; Manna, S.L.; Novellino, E.; Malfitano, A.M.; Marasco, D. Molecular signaling involving intrinsically disordered proteins in prostate cancer. Asian J. Androl. 2016, 18, 673–681. [Google Scholar] [PubMed]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today 2019, 24, 217–227. [Google Scholar] [CrossRef] [PubMed]


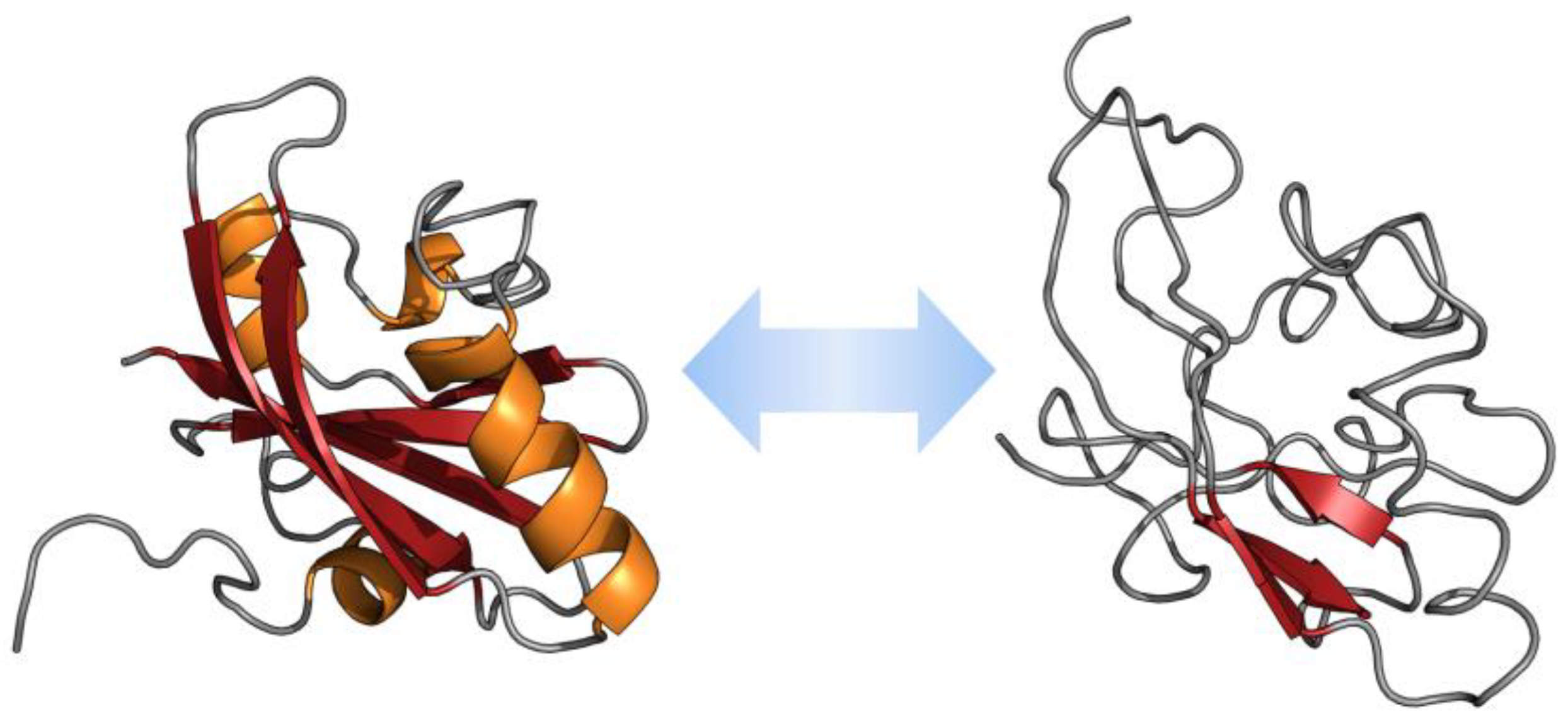{kind=link}
| Compound Name * | Targets | Compound Structure |
|---|---|---|
| CLR01 (Molecular tweezers) | Lysine and arginine residues in amyloid proteins |  |
| ELN484228 | α-Synuclein |  |
| SEN1576 | Amyloid β |  |
| NQTrp | PHF6 (Tau protein) |  |
| Nutlin-3 | p53-MDM2 complex |  |
| 10058-F4 | c-Myc-Max complex |  |
| 10074-G5 | c-Myc-Max complex |  |
| YK-4–279 | EWS-Fli1 |  |
| Trodusquemine | PTP1B |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, A.H.S.; Lopes, F.C.; John, E.B.O.; Carlini, C.R.; Ligabue-Braun, R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int. J. Mol. Sci. 2019, 20, 1322. https://doi.org/10.3390/ijms20061322
Martinelli AHS, Lopes FC, John EBO, Carlini CR, Ligabue-Braun R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. International Journal of Molecular Sciences. 2019; 20(6):1322. https://doi.org/10.3390/ijms20061322
Chicago/Turabian StyleMartinelli, Anne H. S., Fernanda C. Lopes, Elisa B. O. John, Célia R. Carlini, and Rodrigo Ligabue-Braun. 2019. "Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies" International Journal of Molecular Sciences 20, no. 6: 1322. https://doi.org/10.3390/ijms20061322
APA StyleMartinelli, A. H. S., Lopes, F. C., John, E. B. O., Carlini, C. R., & Ligabue-Braun, R. (2019). Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. International Journal of Molecular Sciences, 20(6), 1322. https://doi.org/10.3390/ijms20061322