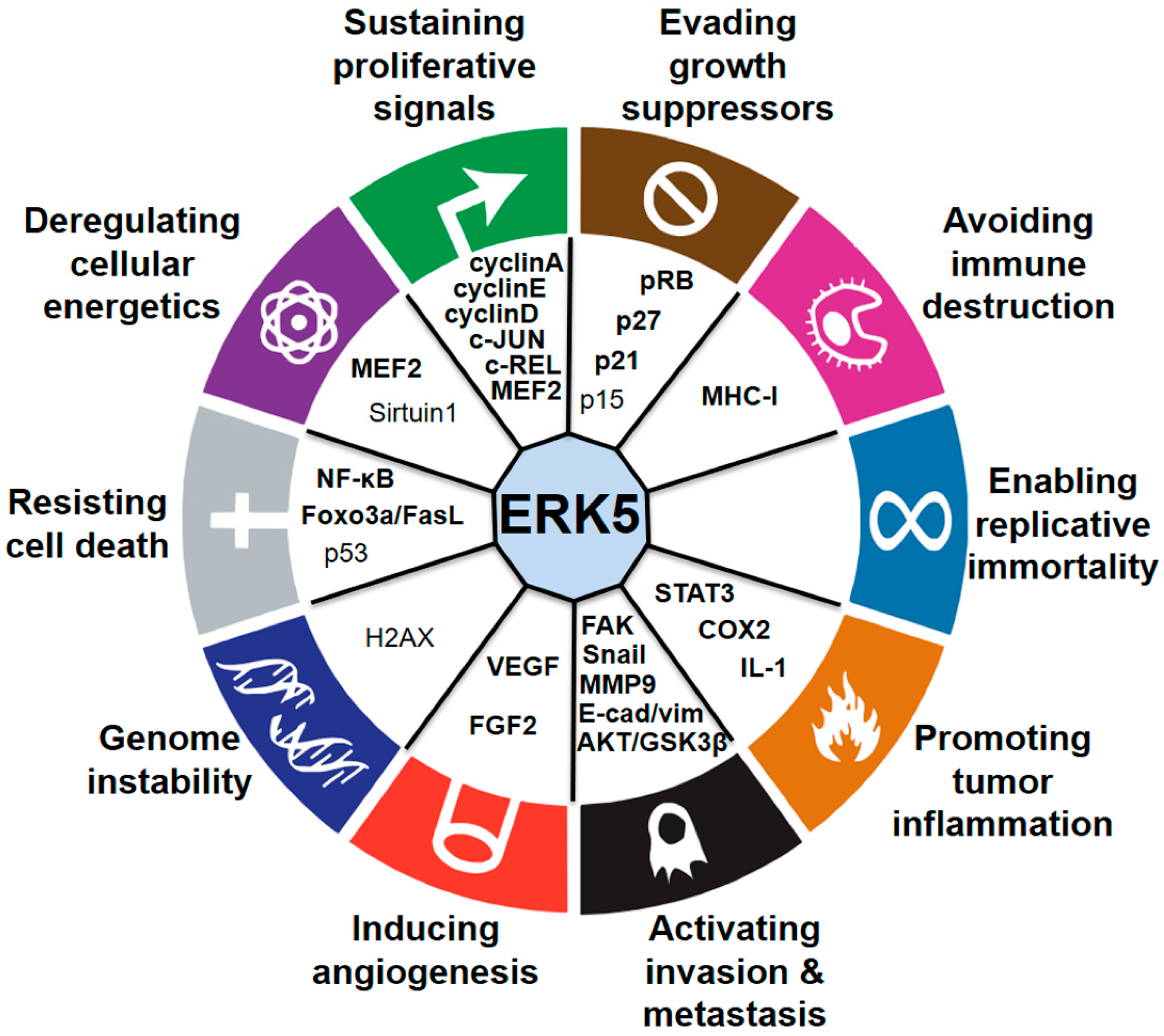
Impact of ERK5 on the Hallmarks of Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
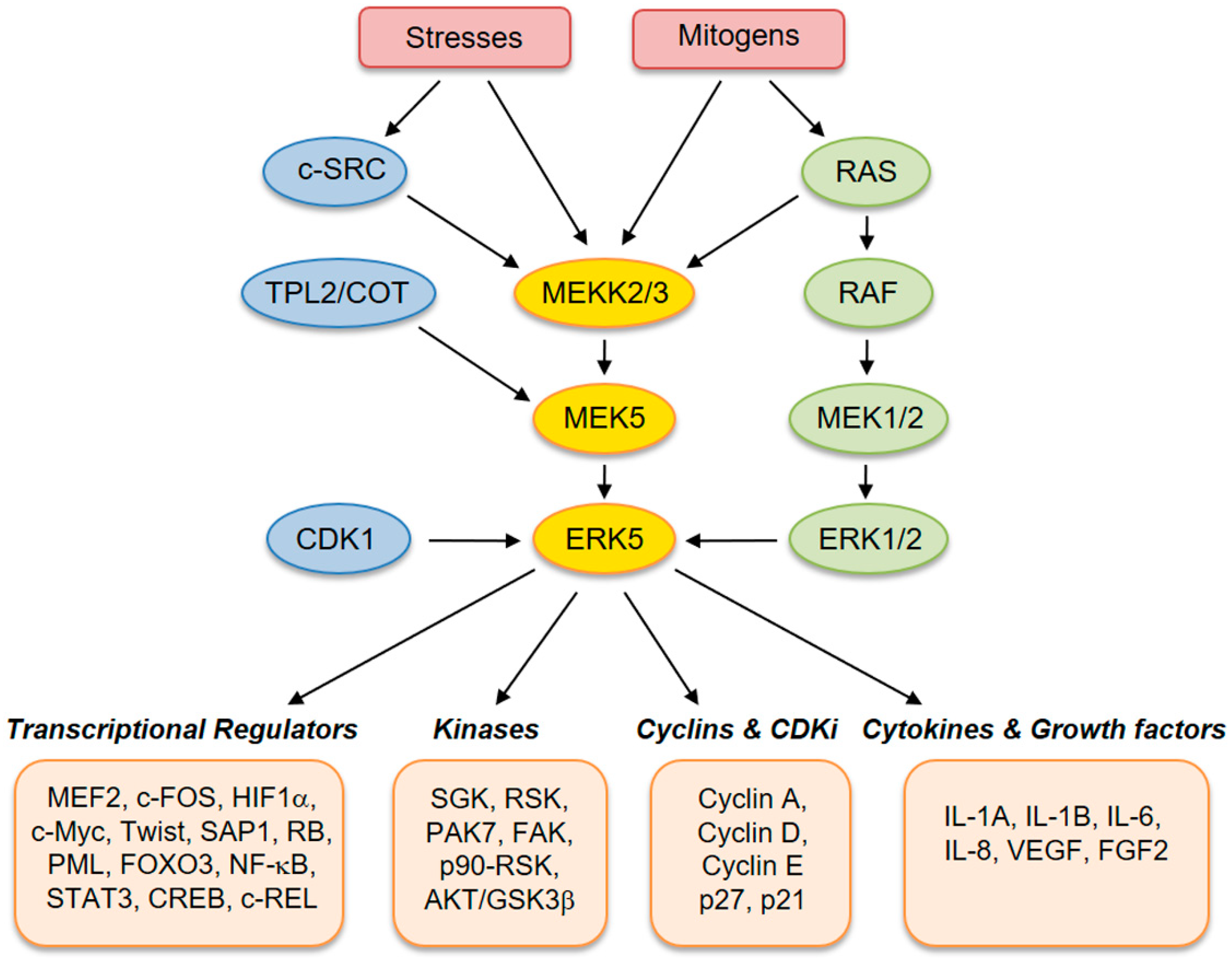
2. The ERK5 Signaling Pathway
3. Sustaining Proliferative Signals
4. Evading Growth Suppressors
5. Avoiding Immune Destruction
6. Enabling Replicative Immortality
7. Promoting Tumor Inflammation
8. Activating Invasion and Metastasis
9. Inducing Angiogenesis
10. Genome Instability
11. Resisting Cell Death
12. Deregulating Cellular Energetics
13. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK | Anaplastic lymphoma kinase |
| AP-1 | Activator protein-1 |
| ATM | Ataxia telangiectasia mutated |
| BC | Breast cancer |
| BMK-1 | Big mitogen-activated protein kinase 1 |
| BNIP3 | BCL2 interacting protein 3 |
| BNIP3L | BNIP3 like |
| CCA | Cholangiocarcinoma |
| CCRC | Clear cell renal carcinoma |
| CDKi | Cyclin-dependent protein kinase inhibitor |
| CML | Chronic myeloid leukemia |
| COX2 | Cyclooxygenase 2 |
| CRC | Colorectal cancer |
| DDIAS | DNA-damage-induced apoptosis suppressor |
| EGF | Epidermal growth factor |
| E-cad | E-cadherin |
| EMT | Epithelial-to-mesenchymal transition |
| ERK5 | Extracellular signal-regulated kinase 5 |
| ERα | Estrogen receptor α |
| FAK | Focal adhesion kinase |
| FGF | Fibroblast growth factor |
| GSK3 | Glycogen synthase kinase 3 |
| HCC | Hepatocellular carcinoma |
| HGF | Hepatocyte growth factor |
| IL | Interleukin |
| MAPK | Mitogen-activated protein kinase family |
| MEF-2 | Myocyte enhancer factor 2 |
| MHC-I | Major histocompatibility complex class I |
| MM | Multiple myeloma |
| MMP | Matrix metalloproteinase |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLS | Nuclear localization signal |
| OS | Osteosarcoma |
| OXPHOS | Oxidative phosphorylation |
| PC | Prostate cancer |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDGF | Platelet-derived growth factor |
| PI3K | PhosphoInositide 3-kinase |
| SATB2 | Special AT-rich sequence-binding protein 2 |
| SGK | Serum/glucocorticoid-regulated kinase |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tumor-associated macrophages |
| TGF-β | Transforming growth factor β |
| TNBC | Triple-negative breast cancer |
| TNFα | Tumor necrosis factor α |
| TβRI | TGF-β receptor type I |
| VEGF | Vascular endothelial growth factor |
| Vim | Vimentin |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.; Kusuhara, M.; Ulevitch, R.J.; Berk, B.C.; Lee, J.D. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 1996, 271, 16586–16590. [Google Scholar] [CrossRef]
- Suzaki, Y.; Yoshizumi, M.; Kagami, S.; Koyama, A.H.; Taketani, Y.; Houchi, H.; Tsuchiya, K.; Takeda, E.; Tamaki, T. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: Potential role in cell survival following oxidative insults. J. Biol. Chem. 2002, 277, 9614–9621. [Google Scholar] [CrossRef]
- Buschbeck, M.; Hofbauer, S.; Di Croce, L.; Keri, G.; Ullrich, A. Abl-kinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep. 2005, 6, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef]
- Morimoto, H.; Kondoh, K.; Nishimoto, S.; Terasawa, K.; Nishida, E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 2007, 282, 35449–35456. [Google Scholar] [CrossRef]
- Mody, N.; Campbell, D.G.; Morrice, N.; Peggie, M.; Cohen, P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 2003, 372, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef]
- Simões, A.E.; Rodrigues, C.M.; Borralho, P.M. The MEK5/ERK5 signalling pathway in cancer: A promising novel therapeutic target. Drug Discov. Today 2016, 21, 1654–1663. [Google Scholar] [CrossRef]
- Barros, J.C.; Marshall, C.J. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J. Cell Sci. 2005, 118, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Müller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell. Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef]
- Hoang, V.T.; Yan, T.J.; Cavanaugh, J.E.; Flaherty, P.T.; Beckman, B.S.; Burow, M.E. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017, 392, 51–59. [Google Scholar] [CrossRef]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [CrossRef]
- Shao, Y.; Akmentin, W.; Toledo-Aral, J.J.; Rosenbaum, J.; Valdez, G.; Cabot, J.B.; Hilbush, B.S.; Halegoua, S. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J. Cell Biol. 2002, 157, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Kesavan, K.; Lobel-Rice, K.; Sun, W.; Lapadat, R.; Webb, S.; Johnson, G.L.; Garrington, T.P. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J. Cell. Physiol. 2004, 199, 140–148. [Google Scholar] [CrossRef]
- Rovida, E.; Spinelli, E.; Sdelci, S.; Barbetti, V.; Morandi, A.; Giuntoli, S.; Dello Sbarba, P. ERK5/BMK1 is indispensable for optimal colony-stimulating factor 1 (CSF-1)-induced proliferation in macrophages in a Src-dependent fashion. J. Immunol. 2008, 180, 4166–4172. [Google Scholar] [CrossRef]
- Rovida, E.; Navari, N.; Caligiuri, A.; Dello Sbarba, P.; Marra, F. ERK5 differentially regulates PDGF-induced proliferation and migration of hepatic stellate cells. J. Hepatol. 2008, 48, 107–115. [Google Scholar] [CrossRef]
- Mulloy, R.; Salinas, S.; Philips, A.; Hipskind, R.A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003, 22, 5387–5398. [Google Scholar] [CrossRef]
- Cude, K.; Wang, Y.; Choi, H.J.; Hsuan, S.L.; Zhang, H.; Wang, C.Y.; Xia, Z. Regulation of the G2-M cell cycle progression by the ERK5-NFkappaB signaling pathway. J. Cell Biol. 2007, 177, 253–264. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, E.; Pandiella, A. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 2010, 123, 3146–3156. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.B.; Jenkins, B.L.; McCarthy, L.; Thilak, L.; Robson, C.N.; Neal, D.E.; Leung, H.Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003, 22, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- McCracken, S.R.; Ramsay, A.; Heer, R.; Mathers, M.E.; Jenkins, B.L.; Edwards, J.; Robson, C.N.; Marquez, R.; Cohen, P.; Leung, H.Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978–2988. [Google Scholar] [CrossRef]
- Ramsay, A.K.; McCracken, S.R.; Soofi, M.; Fleming, J.; Yu, A.X.; Ahmad, I.; Morland, R.; Machesky, L.; Nixon, C.; Edwards, D.R.; et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br. J. Cancer 2011, 104, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Zhang, L.; Wang, T. Phosphorylation of BMK1 induces prostatic carcinoma cell proliferation by promoting entry into the S phase of the cell cycle. Oncol. Lett. 2016, 11, 99–104. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, C.; Ma, X.; Wu, R.; Zhu, W.; Li, X.; Liang, Z.; Deng, F.; Wu, J.; Geng, S.; et al. Phthalates promote prostate cancer cell proliferation through activation of ERK5 and p38. Environ. Toxicol. Pharmacol. 2018, 63, 29–33. [Google Scholar] [CrossRef]
- Ahmad, I.; Singh, L.B.; Yang, Z.H.; Kalna, G.; Fleming, J.; Fisher, G.; Cooper, C.; Cuzick, J.; Berney, D.M.; Møller, H.; et al. Mir143 expression inversely correlates with nuclear ERK5 immunoreactivity in clinical prostate cancer. Br. J. Cancer 2013, 108, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Clapé, C.; Fritz, V.; Henriquet, C.; Apparailly, F.; Fernandez, P.L.; Iborra, F.; Avancès, C.; Villalba, M.; Culine, S.; Fajas, L. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS ONE 2009, 4, e7542. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Maruo, K.; Yamada, N.; Kitade, Y.; Naoe, T.; Akao, Y. MicroRNA-143 functions as a tumor suppressor in human bladder cancer T24 cells. Cancer Lett. 2011, 307, 211–220. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, J.; Luo, S.; Yi, J.; Wang, P.; Zheng, Q.; Wen, Y. miR-143 is associated with proliferation and apoptosis involving ERK5 in HeLa cells. Oncol. Lett. 2016, 12, 3021–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Ma, C.; Li, W.; Yang, S.; Liu, Z. miR-143 suppresses epithelial-mesenchymal transition and inhibits tumor growth of breast cancer through down-regulation of ERK5. Mol. Carcinog. 2016, 55, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Garaude, J.; Cherni, S.; Kaminski, S.; Delepine, E.; Chable-Bessia, C.; Benkirane, M.; Borges, J.; Pandiella, A.; Iñiguez, M.A.; Fresno, M.; et al. ERK5 activates NF-kappaB in leukemic T cells and is essential for their growth in vivo. J. Immunol. 2006, 177, 7607–7617. [Google Scholar] [CrossRef] [PubMed]
- Charni, S.; Aguilo, J.I.; Garaude, J.; de Bettignies, G.; Jacquet, C.; Hipskind, R.A.; Singer, D.; Anel, A.; Villalba, M. ERK5 knockdown generates mouse leukemia cells with low MHC class I levels that activate NK cells and block tumorigenesis. J. Immunol. 2009, 182, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Tusa, I.; Cheloni, G.; Poteti, M.; Gozzini, A.; DeSouza, N.H.; Shan, Y.; Deng, X.; Gray, N.S.; Li, S.; Rovida, E.; et al. Targeting the Extracellular Signal-Regulated Kinase 5 Pathway to Suppress Human Chronic Myeloid Leukemia Stem Cells. Stem Cell Rep. 2018, 11, 929–943. [Google Scholar] [CrossRef]
- Marzi, I.; Cipolleschi, M.G.; D’Amico, M.; Stivarou, T.; Rovida, E.; Vinci, M.C.; Pandolfi, S.; Dello Sbarba, P.; Stecca, B.; Olivotto, M. The involvement of a Nanog, Klf4 and c-Myc transcriptional circuitry in the intertwining between neoplastic progression and reprogramming. Cell Cycle 2013, 12, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madak-Erdogan, Z.; Ventrella, R.; Petry, L.; Katzenellenbogen, B.S. Novel roles for ERK5 and cofilin as critical mediators linking ERα-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol. Cancer Res. 2014, 12, 714–727. [Google Scholar] [CrossRef]
- Ortiz-Ruiz, M.J.; Álvarez-Fernández, S.; Parrott, T.; Zaknoen, S.; Burrows, F.J.; Ocaña, A.; Pandiella, A.; Esparís-Ogando, A. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget 2014, 5, 11308–11318. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Fernández, S.; Ortiz-Ruiz, M.J.; Parrott, T.; Zaknoen, S.; Ocio, E.M.; San Miguel, J.; Burrows, F.J.; Esparís-Ogando, A.; Pandiella, A. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clin. Cancer Res. 2013, 19, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Miranda, M.; Shi, W.; Simpson, P.T.; Song, S.; Vargas, A.C.; Saunus, J.M.; Smart, C.E.; Mariasegaram, M.; Wiegmans, A.P.; et al. Kinome profiling reveals breast cancer heterogeneity and identifies targeted therapeutic opportunities for triple negative breast cancer. Oncotarget 2014, 5, 3145–3158. [Google Scholar] [CrossRef] [Green Version]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodríguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gómez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lee, H.; Park, Y.S.; Lee, Y.; Seo, S.W. ERK5 regulates invasiveness of osteosarcoma by inducing MMP-9. J. Orthop. Res. 2012, 30, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Lv, B.; Li, Y.; Cheng, Q.; Su, C.; Yin, G. MiR-143 regulates the proliferation and migration of osteosarcoma cells through targeting MAPK7. Arch. Biochem. Biophys. 2017, 630, 47–53. [Google Scholar] [CrossRef]
- Carvajal-Vergara, X.; Tabera, S.; Montero, J.C.; Esparís-Ogando, A.; López-Pérez, R.; Mateo, G.; Gutiérrez, N.; Parmo-Cabañas, M.; Teixidó, J.; San Miguel, J.F.; et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, P.R.; Taniguchi, K.; Harris, A.R.; Bertin, S.; Takahashi, N.; Duong, J.; Campos, A.D.; Powis, G.; Corr, M.; Karin, M.; et al. ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation. Nat. Commun. 2016, 7, 11551. [Google Scholar] [CrossRef] [Green Version]
- Simões, A.E.; Pereira, D.M.; Gomes, S.E.; Brito, H.; Carvalho, T.; French, A.; Castro, R.E.; Steer, C.J.; Thibodeau, S.N.; Rodrigues, C.M.; et al. Aberrant MEK5/ERK5 signalling contributes to human colon cancer progression via NF-κB activation. Cell Death Dis. 2015, 6, e1718. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.M.; Simões, A.E.; Gomes, S.E.; Castro, R.E.; Carvalho, T.; Rodrigues, C.M.; Borralho, P.M. MEK5/ERK5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget 2016, 7, 34322–34340. [Google Scholar] [CrossRef] [Green Version]
- Tatake, R.J.; O’Neill, M.M.; Kennedy, C.A.; Wayne, A.L.; Jakes, S.; Wu, D.; Kugler, S.Z., Jr.; Kashem, M.A.; Kaplita, P.; Snow, R.J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Clark, J.; Wang, L.Z.; Gilmour, L.; Squires, M.; Gilley, R.; Foxton, C.; Newell, D.R.; Wedge, S.R.; Cook, S.J. Tumor cells with KRAS or BRAF mutations or ERK5/MAPK7 amplification are not addicted to ERK5 activity for cell proliferation. Cell Cycle 2016, 15, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, K.; Zhang, J.; Xiong, M.; Wang, X.; Luo, X.; Han, L.; Meng, Y.; Zhang, Y.; Liao, W.; Liu, S. CDK5 functions as a tumor promoter in human colorectal cancer via modulating the ERK5-AP-1 axis. Cell Death Dis. 2016, 7, e2415. [Google Scholar] [CrossRef]
- Borralho, P.M.; Simões, A.E.; Gomes, S.E.; Lima, R.T.; Carvalho, T.; Ferreira, D.M.; Vasconcelos, M.H.; Castro, R.E.; Rodrigues, C.M. miR-143 overexpression impairs growth of human colon carcinoma xenografts in mice with induction of apoptosis and inhibition of proliferation. PLoS ONE 2011, 6, e23787. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grünweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.A.; Hyodo, T.; Ito, S.; Kurita, K.; Kokuryo, T.; Uehara, K.; Nagino, M.; Takahashi, M.; Hamaguchi, M.; Senga, T. SATB2 suppresses the progression of colorectal cancer cells via inactivation of MEK5/ERK5 signaling. FEBS J. 2015, 282, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, J.; Qin, Y.; Mo, X.; Huang, M.; Ru, H.; Yang, Y.; Liu, J.; Lin, Y. SATB2 suppresses gastric cancer cell proliferation and migration. Tumour Biol. 2016, 37, 4597–4602. [Google Scholar] [CrossRef] [PubMed]
- Arias-González, L.; Moreno-Gimeno, I.; del Campo, A.R.; Serrano-Oviedo, L.; Valero, M.L.; Esparís-Ogando, A.; de la Cruz-Morcillo, M.Á.; Melgar-Rojas, P.; García-Cano, J.; Cimas, F.J.; et al. ERK5/BMK1 is a novel target of the tumor suppressor VHL: Implication in clear cell renal carcinoma. Neoplasia 2013, 15, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Nino, M.E.; Blumen, S.R.; Sabo-Attwood, T.; Pass, H.; Carbone, M.; Testa, J.R.; Altomare, D.A.; Mossman, B.T. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am. J. Respir. Cell Mol. Biol. 2008, 38, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Miller, J.M.; Cason, C.; Sayan, M.; MacPherson, M.B.; Beuschel, S.L.; Hillegass, J.; Vacek, P.M.; Pass, H.I.; Mossman, B.T. Extracellular signal-regulated kinase 5: A potential therapeutic target for malignant mesotheliomas. Clin. Cancer Res. 2013, 19, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Sureban, S.M.; May, R.; Weygant, N.; Qu, D.; Chandrakesan, P.; Bannerman-Menson, E.; Ali, N.; Pantazis, P.; Westphalen, C.B.; Wang, T.C.; et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014, 351, 151–161. [Google Scholar] [CrossRef]
- Umapathy, G.; El Wakil, A.; Witek, B.; Chesler, L.; Danielson, L.; Deng, X.; Gray, N.S.; Johansson, M.; Kvarnbrink, S.; Ruuth, K.; et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci. Signal. 2014, 7, ra102. [Google Scholar] [CrossRef]
- Zen, K.; Yasui, K.; Nakajima, T.; Zen, Y.; Zen, K.; Gen, Y.; Mitsuyoshi, H.; Minami, M.; Mitsufuji, S.; Tanaka, S.; et al. ERK5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes Chromosomes Cancer. 2009, 48, 109–120. [Google Scholar] [CrossRef]
- Zamani, A.; Fan, H.; Luo, G. Identification of cellular genes and pathways important for tumorigenicity of hepatocellular carcinoma cell lines by proteomic profiling. Oncotarget 2017, 8, 96171–96183. [Google Scholar] [CrossRef] [Green Version]
- Rovida, E.; Di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparís-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Urso, C.; Borgognoni, L.; Wang, J.; Deng, X.; Gray, N.S.; Stecca, B.; et al. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene 2018, 37, 2601–2614. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wang, L.; Xu, Q.; Wang, K.; Xie, D.; Yu, Z.; Jiang, K.; Liao, L.; Yates, J.R.; Lee, J.D.; et al. Targeting BMK1 Impairs the Drug Resistance to Combined Inhibition of BRAF and MEK1/2 in Melanoma. Sci. Rep. 2017, 7, 46244. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Fdez, A.; Ortiz-Ruiz, M.J.; Re-Louhau, M.F.; Ramos, I.; Blanco-Múñez, Ó.; Ludeña, D.; Abad, M.; Sánchez-Martín, M.; Pandiella, A.; Esparís-Ogando, A. MEK5 promotes lung adenocarcinoma. Eur. Respir. J. 2018. [Google Scholar] [CrossRef]
- Perez-Madrigal, D.; Finegan, K.G.; Paramo, B.; Tournier, C. The extracellular-regulated protein kinase 5 (ERK5) promotes cell proliferation through the down-regulation of inhibitors of cyclin dependent protein kinases (CDKs). Cell. Signal. 2012, 24, 2360–2368. [Google Scholar] [CrossRef]
- Wang, X.; Gocek, E.; Novik, V.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. Inhibition of Cot1/Tlp2 oncogene in AML cells reduces ERK5 activation and up-regulates p27Kip1 concomitant with enhancement of differentiation and cell cycle arrest induced by silibinin and 1,25-dihydroxyvitamin D(3). Cell Cycle 2010, 9, 4542–4551. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., 3rd; Gray, N.S.; Lee, J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef]
- Yang, Q.; Liao, L.; Deng, X.; Chen, R.; Gray, N.S.; Yates, J.R., 3rd; Lee, J.D. BMK1 is involved in the regulation of p53 through disrupting the PML-MDM2 interaction. Oncogene 2013, 32, 3156–3164. [Google Scholar] [CrossRef]
- Granados-Jaén, A.; Angulo-Ibáñez, M.; Rovira-Clavé, X.; Gamez, C.P.; Soriano, F.X.; Reina, M.; Espel, E. Absence of ERK5/MAPK7 delays tumorigenesis in Atm−/− mice. Oncotarget 2016, 7, 74435–74447. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wang, P.; Li, X.; Wang, Q.; Deng, Z.B.; Zhuang, X.; Mu, J.; Zhang, L.; Wang, B.; Yan, J.; et al. Restoration of miR17/20a in solid tumor cells enhances the natural killer cell antitumor activity by targeting Mekk2. Cancer Immunol. Res. 2014, 2, 789–799. [Google Scholar] [CrossRef]
- Loveridge, C.J.; Mui, E.J.; Patel, R.; Tan, E.H.; Ahmad, I.; Welsh, M.; Galbraith, J.; Hedley, A.; Nixon, C.; Blyth, K.; et al. Increased T-cell Infiltration Elicited by Erk5 Deletion in a Pten-Deficient Mouse Model of Prostate Carcinogenesis. Cancer Res. 2017, 77, 3158–3168. [Google Scholar] [CrossRef]
- Uziel, O.; Yosef, N.; Sharan, R.; Ruppin, E.; Kupiec, M.; Kushnir, M.; Beery, E.; Cohen-Diker, T.; Nordenberg, J.; Lahav, M. The effects of telomere shortening on cancer cells: A network model of proteomic and microRNA analysis. Genomics 2015, 105, 5–16. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Dabrowska, M.; Skoneczny, M.; Rode, W. Functional gene expression profile underlying methotrexate-induced senescence in human colon cancer cells. Tumour Biol. 2011, 32, 965–976. [Google Scholar] [CrossRef]
- Finegan, K.G.; Perez-Madrigal, D.; Hitchin, J.R.; Davies, C.C.; Jordan, A.M.; Tournier, C. ERK5 is a critical mediator of inflammation-driven cancer. Cancer Res. 2015, 75, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.K.; Shukla, A.; Leggett, A.L.; Munson, P.B.; Miller, J.M.; MacPherson, M.B.; Beuschel, S.L.; Pass, H.I.; Shukla, A. Extracellular signal regulated kinase 5 and inflammasome in progression of mesothelioma. Oncotarget 2017, 9, 293–305. [Google Scholar] [CrossRef]
- Wang, X.; Pesakhov, S.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. ERK5 pathway regulates transcription factors important for monocytic differentiation of human myeloid leukemia cells. J. Cell. Physiol. 2014, 229, 856–867. [Google Scholar] [CrossRef]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Villa-Moruzzi, E. Targeting of FAK Ser910 by ERK5 and PP1delta in non-stimulated and phorbol ester-stimulated cells. Biochem. J. 2007, 408, 7–18. [Google Scholar] [CrossRef]
- Villa-Moruzzi, E. Tyrosine phosphatases in the HER2-directed motility of ovarian cancer cells: Involvement of PTPN12, ERK5 and FAK. Anal. Cell. Pathol. (Amst.) 2011, 34, 101–112. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Xu, S.; Wang, S.; Tu, Y.; Xiong, Y.; Mei, J.; Wang, C. The role of MAPK signaling pathway in the Her-2-positive meningiomas. Oncol. Rep. 2016, 36, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Wang, T.; Wang, W.; Zhang, S.; Liao, Y.; Chen, J. Role of MAPK7 in cell proliferation and metastasis in ovarian cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 10444–10451. [Google Scholar]
- Sawhney, R.S.; Liu, W.; Brattain, M.G. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J. Cell. Physiol. 2009, 219, 152–161. [Google Scholar] [CrossRef]
- Castro, N.E.; Lange, C.A. Breast tumor kinase and extracellular signal-regulated kinase 5 mediate Met receptor signaling to cell migration in breast cancer cells. Breast Cancer Res. 2010, 12, R60. [Google Scholar] [CrossRef]
- Locatelli, A.; Lange, C.A. Met receptors induce Sam68-dependent cell migration by activation of alternate extracellular signal-regulated kinase family members. J. Biol. Chem. 2011, 286, 21062–21072. [Google Scholar] [CrossRef]
- Cronan, M.R.; Nakamura, K.; Johnson, N.L.; Granger, D.A.; Cuevas, B.D.; Wang, J.G.; Mackman, N.; Scott, J.E.; Dohlman, H.G.; Johnson, G.L. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene 2012, 31, 3889–3900. [Google Scholar] [CrossRef]
- Mirza, A.A.; Kahle, M.P.; Ameka, M.; Campbell, E.M.; Cuevas, B.D. MEKK2 regulates focal adhesion stability and motility in invasive breast cancer cells. Biochim. Biophys. Acta 2014, 1843, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Im, J.Y.; Yoon, S.H.; Kim, B.K.; Ban, H.S.; Won, K.J.; Chungm, K.S.; Jung, K.E.; Won, M. DNA damage induced apoptosis suppressor (DDIAS) is upregulated via ERK5/MEF2B signaling and promotes β-catenin-mediated invasion. Biochim. Biophys. Acta 2016, 1859, 1449–1458. [Google Scholar] [CrossRef]
- Zuo, Y.; Wu, Y.; Wehrli, B.; Chakrabarti, S.; Chakraborty, C. Modulation of ERK5 is a novel mechanism by which Cdc42 regulates migration of breast cancer cells. J. Cell. Biochem. 2015, 116, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Asara, J.M.; Tyner, A.L. Protein-tyrosine kinase 6 promotes peripheral adhesion complex formation and cell migration by phosphorylating p130 CRK-associated substrate. J. Biol. Chem. 2012, 287, 148–158. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, B.; Guo, W.; Gao, L.; Shi, L.; Li, H.; Lu, S.; Liu, Y.; Li, X. miR-429 inhibits glioma invasion through BMK1 suppression. J. Neurooncol. 2015, 125, 43–54. [Google Scholar] [CrossRef]
- Wu, J.; Cui, H.; Zhu, Z.; Wang, L. MicroRNA-200b-3p suppresses epithelial-mesenchymal transition and inhibits tumor growth of glioma through down-regulation of ERK5. Biochem. Biophys. Res. Commun. 2016, 478, 1158–1164. [Google Scholar] [CrossRef]
- Zhang, D.; Li, H.; Jiang, X.; Cao, L.; Wen, Z.; Yang, X.; Xue, P. Role of AP-2α and MAPK7 in the regulation of autocrine TGF-β/miR-200b signals to maintain epithelial-mesenchymal transition in cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesser-Gamba, F.; Petrilli, A.S.; de Seixas Alves, M.T.; Filho, R.J.; Juliano, Y.; Toledo, S.R. MAPK7 and MAP2K4 as prognostic markers in osteosarcoma. Hum. Pathol. 2012, 43, 994–1002. [Google Scholar] [CrossRef]
- Yue, B.; Ren, Q.X.; Su, T.; Wang, L.N.; Zhang, L. ERK5 silencing inhibits invasion of human osteosarcoma cell via modulating the Slug/MMP-9 pathway. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2640–2647. [Google Scholar]
- Huang, Y.; Yao, J.; Zhu, B.; Zhang, J.; Sun, T. Mitogen-activated protein kinase 7 promotes cell proliferation, migration and invasion in SOSP-M human osteosarcoma cell line. Tumori 2017, 103, 483–488. [Google Scholar] [CrossRef]
- Tesser-Gamba, F.; Lopes, L.J.; Petrilli, A.S.; Toledo, S.R. MAPK7 gene controls proliferation, migration and cell invasion in osteosarcoma. Mol. Carcinog. 2016, 55, 1700–1713. [Google Scholar] [CrossRef]
- Sticht, C.; Freier, K.; Knöpfle, K.; Flechtenmacher, C.; Pungs, S.; Hofele, C.; Hahn, M.; Joos, S.; Lichter, P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC). Neoplasia 2008, 10, 462–470. [Google Scholar] [CrossRef]
- Serrano-Oviedo, L.; Giménez-Bachs, J.M.; Nam-Cha, S.Y.; Cimas, F.J.; García-Cano, J.; Sánchez-Prieto, R.; Salinas-Sánchez, A.S. Implication of VHL, ERK5, and HIF-1alpha in clear cell renal cell carcinoma: Molecular basis. Urol. Oncol. 2017, 35, e15–e22. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Zhou, C.; Nitschke, A.M.; Xiong, W.; Zhang, Q.; Tang, Y.; Bloch, M.; Elliott, S.; Zhu, Y.; Bazzone, L.; Yu, D.; et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008, 10, R105. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Naoe, T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol. Rep. 2006, 16, 845–850. [Google Scholar] [CrossRef]
- Xia, C.; Yang, Y.; Kong, F.; Kong, Q.; Shan, C. MiR-143-3p inhibits the proliferation, cell migration and invasion of human breast cancer cells by modulating the expression of MAPK7. Biochimie 2018, 147, 98–104. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, H.; Song, H. Upregulation of MEK5 by Stat3 promotes breast cancer cell invasion and metastasis. Oncol. Rep. 2017, 37, 83–90. [Google Scholar] [CrossRef]
- Javaid, S.; Zhang, J.; Smolen, G.A.; Yu, M.; Wittner, B.S.; Singh, A.; Arora, K.S.; Madden, M.W.; Desai, R.; Zubrowski, M.J.; et al. MAPK7 Regulates EMT Features and Modulates the Generation of CTCs. Mol. Cancer Res. 2015, 13, 934–943. [Google Scholar] [CrossRef]
- Pavan, S.; Meyer-Schaller, N.; Diepenbruck, M.; Kalathur, R.K.R.; Saxena, M.; Christofori, G. A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene 2018, 37, 4197–4213. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, T.; Deng, Q.; Zhou, Q.; Sun, X.; Li, E.; Yu, D.; Zhong, C. Benzidine Induces Epithelial-Mesenchymal Transition of Human Bladder Cancer Cells through Activation of ERK5 Pathway. Mol. Cells 2018, 41, 188–197. [Google Scholar] [CrossRef]
- Park, S.J.; Choi, Y.S.; Lee, S.; Lee, Y.J.; Hong, S.; Han, S.; Kim, B.C. BIX02189 inhibits TGF-β1-induced lung cancer cell metastasis by directly targeting TGF-β type I receptor. Cancer Lett. 2016, 381, 314–322. [Google Scholar] [CrossRef]
- Chen, R.; Yang, Q.; Lee, J.D. BMK1 kinase suppresses epithelial-mesenchymal transition through the Akt/GSK3β signaling pathway. Cancer Res. 2012, 72, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Kim, S.W.; Imanaka-Yoshida, K.; Yoshida, T.; Abel, E.D.; Eliceiri, B.; Yang, Y.; Ulevitch, R.J.; Lee, J.D. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Investig. 2004, 113, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Müller, J.; Cross, M.J. The role of ERK5 in endothelial cell function. Biochem. Soc. Trans. 2014, 42, 1584–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Fearns, C.; Eliceiri, B.; Yang, Y.; Lee, J.D. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005, 65, 7699–7706. [Google Scholar] [CrossRef] [PubMed]
- Weldon, C.B.; Scandurro, A.B.; Rolfe, K.W.; Clayton, J.L.; Elliott, S.; Butler, N.N.; Melnik, L.I.; Alam, J.; McLachlan, J.A.; Jaffe, B.M.; et al. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery 2002, 132, 293–301. [Google Scholar] [CrossRef]
- Borges, J.; Pandiella, A.; Esparís-Ogando, A. Erk5 nuclear location is independent on dual phosphorylation, and favours resistance to TRAIL-induced apoptosis. Cell. Signal. 2007, 19, 1473–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Cao, C.; Gong, X.; Rong, L. Inhibition of ERK5 enhances cytarabine-induced apoptosis in acute myeloid leukemia cells. Int. J. Clin. Exp. Med. 2015, 8, 6446–6455. [Google Scholar]
- Razumovskaya, E.; Sun, J.; Rönnstrand, L. Inhibition of MEK5 by BIX02188 induces apoptosis in cells expressing the oncogenic mutant FLT3-ITD. Biochem. Biophys. Res. Commun. 2011, 412, 307–312. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Wang, C.; Xiong, Q.; Lin, Y.; Sun, Q.; Jin, H.; Yang, F.; Ren, X.; Pang, T. Knock-down of CIAPIN1 sensitizes K562 chronic myeloid leukemia cells to Imatinib by regulation of cell cycle and apoptosis-associated members via NF-κB and ERK5 signaling pathway. Biochem. Pharmacol. 2016, 99, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Mo, B.; Hu, C.; Liu, H.; Qi, H.; Wang, X.; Xu, J. Genistein induces cell apoptosis in MDA-MB-231 breast cancer cells via the mitogen-activated protein kinase pathway. Toxicol. In Vitro 2008, 22, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Iio, A.; Naoe, T. Role of microRNA-143 in Fas-mediated apoptosis in human T-cell leukemia Jurkat cells. Leuk. Res. 2009, 33, 1530–1538. [Google Scholar] [CrossRef]
- Wang, X.; Finegan, K.G.; Robinson, A.C.; Knowles, L.; Khosravi-Far, R.; Hinchliffe, K.A.; Boot-Handford, R.P.; Tournier, C. Activation of extracellular signal-regulated protein kinase 5 downregulates FasL upon osmotic stress. Cell Death Differ. 2006, 13, 2099–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Xu, Q.; Jiang, K.; Zhou, G.; Yu, X.; Wang, L.; Zhu, Y.; Fang, L.; Yu, Z.; Lee, J.D.; et al. Inhibition of BMK1 pathway suppresses cancer stem cells through BNIP3 and BNIP3L. Oncotarget 2015, 6, 33279–33289. [Google Scholar] [CrossRef] [Green Version]
- Sturla, L.M.; Cowan, C.W.; Guenther, L.; Castellino, R.C.; Kim, J.Y.; Pomeroy, S.L. A novel role for extracellular signal-regulated kinase 5 and myocyte enhancer factor 2 in medulloblastoma cell death. Cancer Res. 2005, 65, 5683–5689. [Google Scholar] [CrossRef]
- Charni, S.; de Bettignies, G.; Rathore, M.G.; Aguiló, J.I.; van den Elsen, P.J.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J. Immunol. 2010, 185, 3498–3503. [Google Scholar] [CrossRef]
- Lopez-Royuela, N.; Rathore, M.G.; Allende-Vega, N.; Annicotte, J.S.; Fajas, L.; Ramachandran, B.; Gulick, T.; Villalba, M. Extracellular-signal-regulated kinase 5 modulates the antioxidant response by transcriptionally controlling Sirtuin 1 expression in leukemic cells. Int. J. Biochem. Cell Biol. 2014, 53, 253–261. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Rathore, M.G.; Allende-Vega, N.; Vo, D.N.; Belkhala, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; Cartron, G.; Lecellier, C.H.; et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine 2015, 3, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci. Rep. 2018, 8, 7420. [Google Scholar] [CrossRef]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Muñoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat. Commun. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Lee, J.D. Targeting the BMK1 MAP kinase pathway in cancer therapy. Clin. Cancer Res. 2011, 17, 3527–3532. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Erazo, T.; Ferguson, F.M.; Buckley, D.L.; Gomez, N.; Muñoz-Guardiola, P.; Diéguez-Martínez, N.; Deng, X.; Hao, M.; Massefski, W.; et al. Structural and Atropisomeric Factors Governing the Selectivity of Pyrimido-benzodiazipinones as Inhibitors of Kinases and Bromodomains. ACS Chem. Biol. 2018, 13, 2438–2448. [Google Scholar] [CrossRef]
- Myers, S.M.; Bawn, R.H.; Bisset, L.C.; Blackburn, T.J.; Cottyn, B.; Molyneux, L.; Wong, A.C.; Cano, C.; Clegg, W.; Harrington, R.W.; et al. High-Throughput Screening and Hit Validation of Extracellular-Related Kinase 5 (ERK5) Inhibitors. ACS Comb. Sci. 2016, 18, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Elkins, J.M.; Zhang, J.; Yang, Q.; Erazo, T.; Gomez, N.; Choi, H.G.; Wang, J.; Dzamko, N.; Lee, J.D.; et al. Structural determinants for ERK5 (MAPK7) and leucine rich repeat kinase 2 activities of benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-ones. Eur. J. Med. Chem. 2013, 70, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.; Lemos, C.; Wortmann, L.; Eis, K.; Holton, S.J.; Boemer, U.; Moosmayer, D.; Eberspaecher, U.; Weiske, J.; Lechner, C.; et al. Discovery and Characterization of the Potent and Highly Selective (Piperidin-4-yl)pyrido[3,2-d]pyrimidine based in vitro Probe BAY-885 for the Kinase ERK5. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.A.; Fernandez-Alonso, R.; Wang, J.; Toth, R.; Gray, N.S.; Findlay, G.M. Erk5 Is a Key Regulator of Naive-Primed Transition and Embryonic Stem Cell Identity. Cell Rep. 2016, 16, 1820–1828. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.C.; Amantea, C.M.; Nomanbhoy, T.K.; Weissig, H.; Ishiyama, J.; Hu, Y.; Sidique, S.; Li, B.; Kozarich, J.W.; Rosenblum, J.S. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 11865–11870. [Google Scholar] [CrossRef] [Green Version]
- Buschbeck, M.; Ullrich, A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 2005, 280, 2659–2667. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. https://doi.org/10.3390/ijms20061426
Stecca B, Rovida E. Impact of ERK5 on the Hallmarks of Cancer. International Journal of Molecular Sciences. 2019; 20(6):1426. https://doi.org/10.3390/ijms20061426
Chicago/Turabian StyleStecca, Barbara, and Elisabetta Rovida. 2019. "Impact of ERK5 on the Hallmarks of Cancer" International Journal of Molecular Sciences 20, no. 6: 1426. https://doi.org/10.3390/ijms20061426
APA StyleStecca, B., & Rovida, E. (2019). Impact of ERK5 on the Hallmarks of Cancer. International Journal of Molecular Sciences, 20(6), 1426. https://doi.org/10.3390/ijms20061426