The Influence of Trehalose on Atherosclerosis and Hepatic Steatosis in Apolipoprotein E Knockout Mice

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
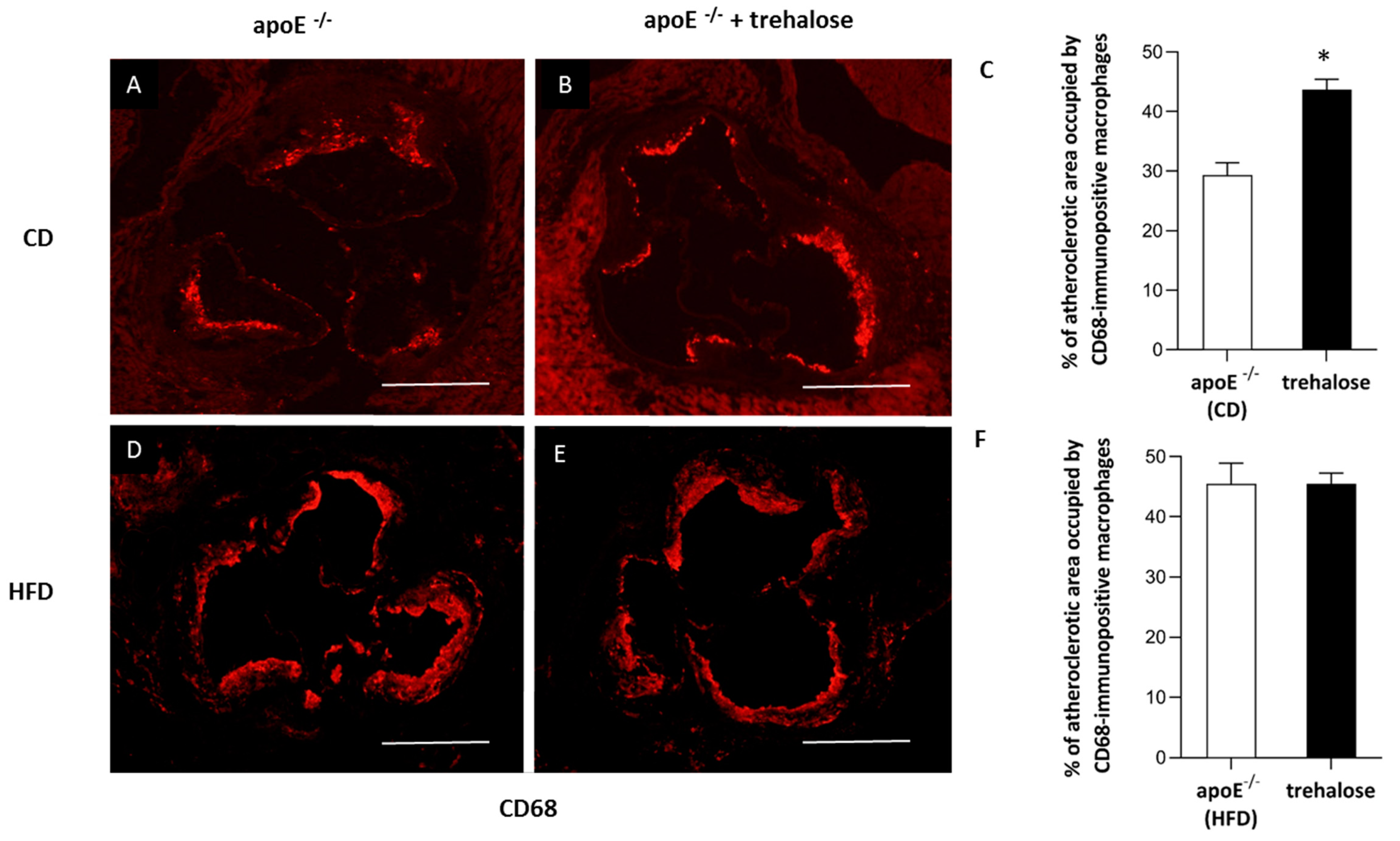
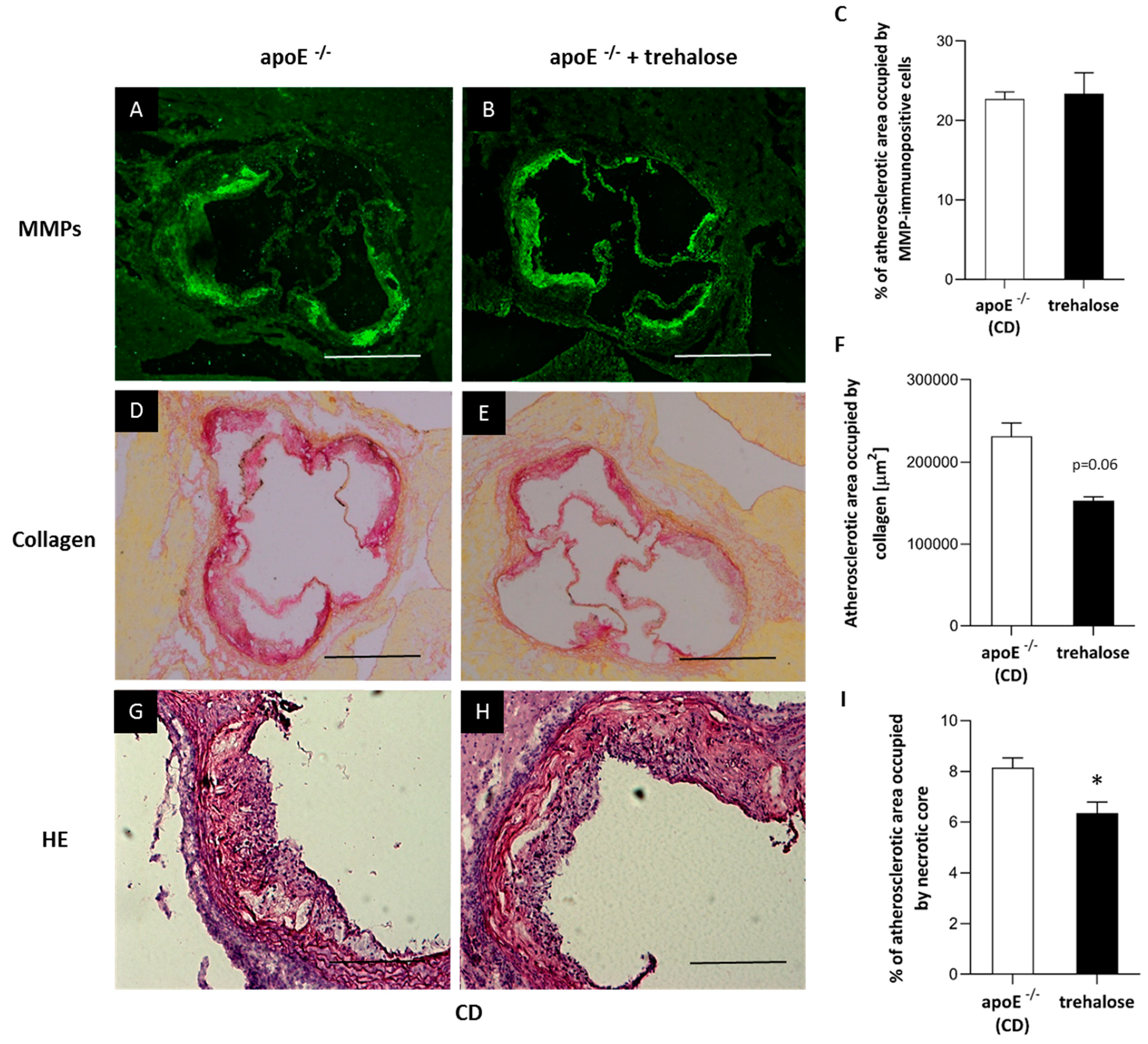
2.1. The Influence of Trehalose on Atherosclerosis
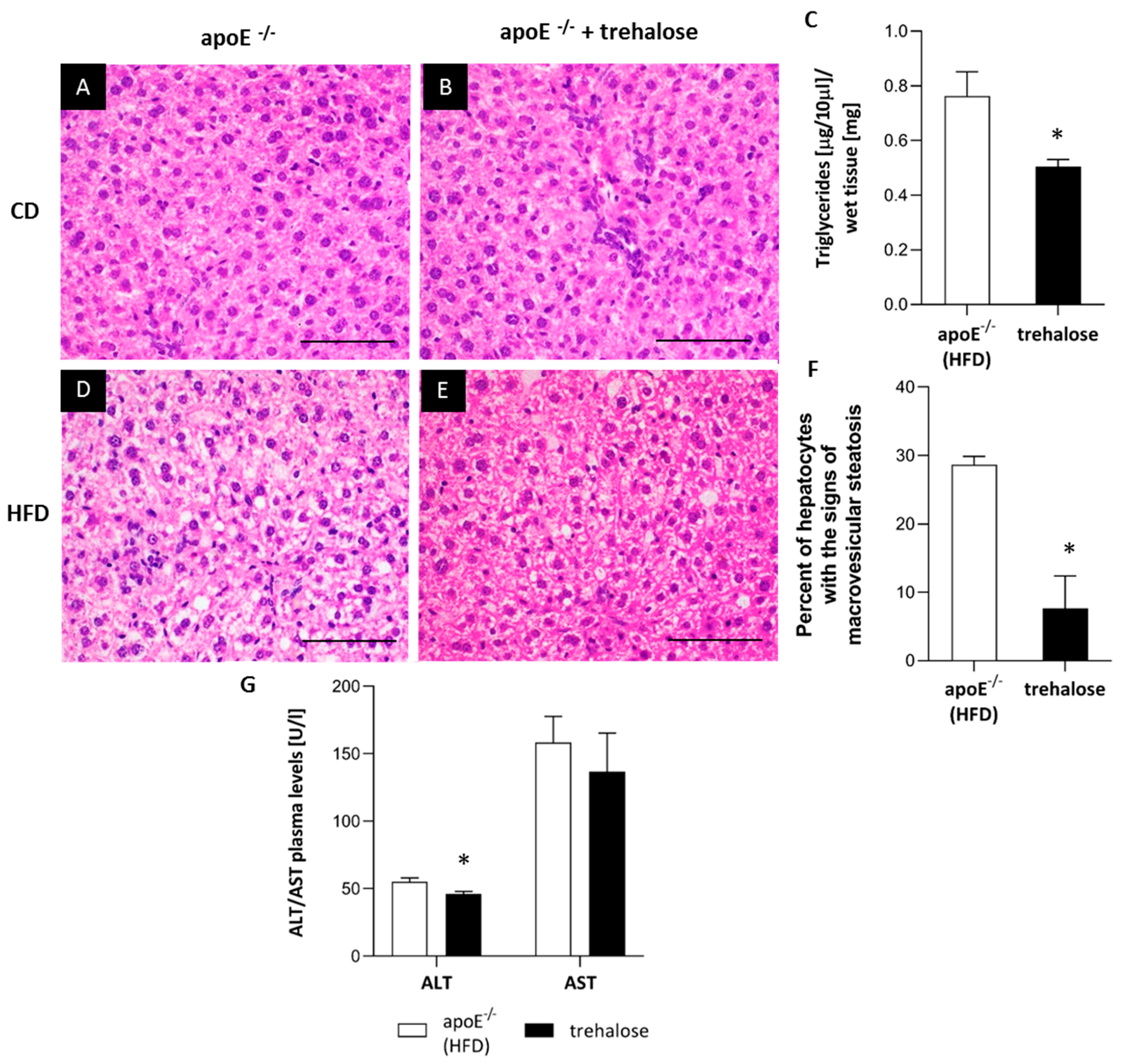
2.2. The Influence of Trehalose on Hepatic Steatosis
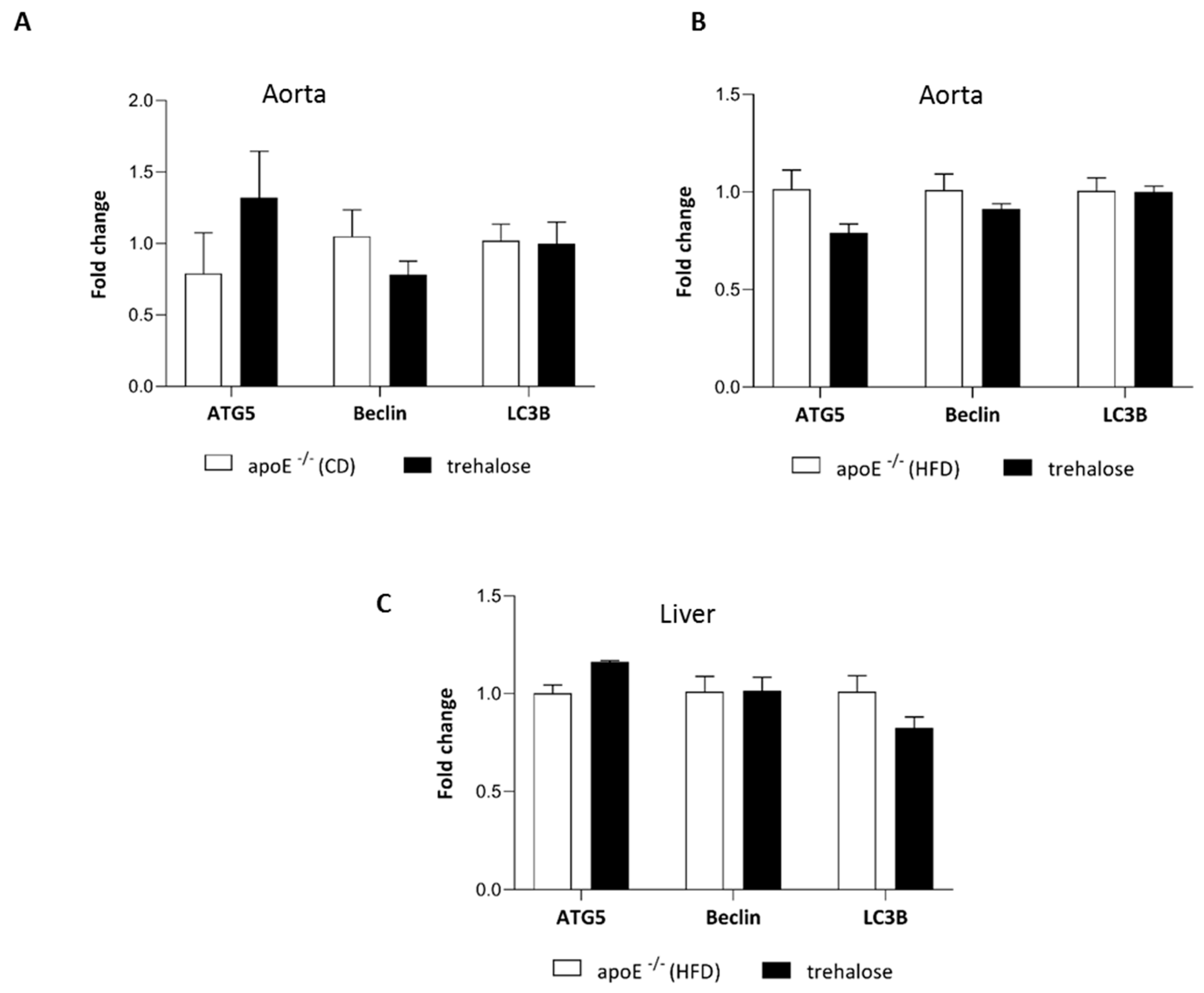
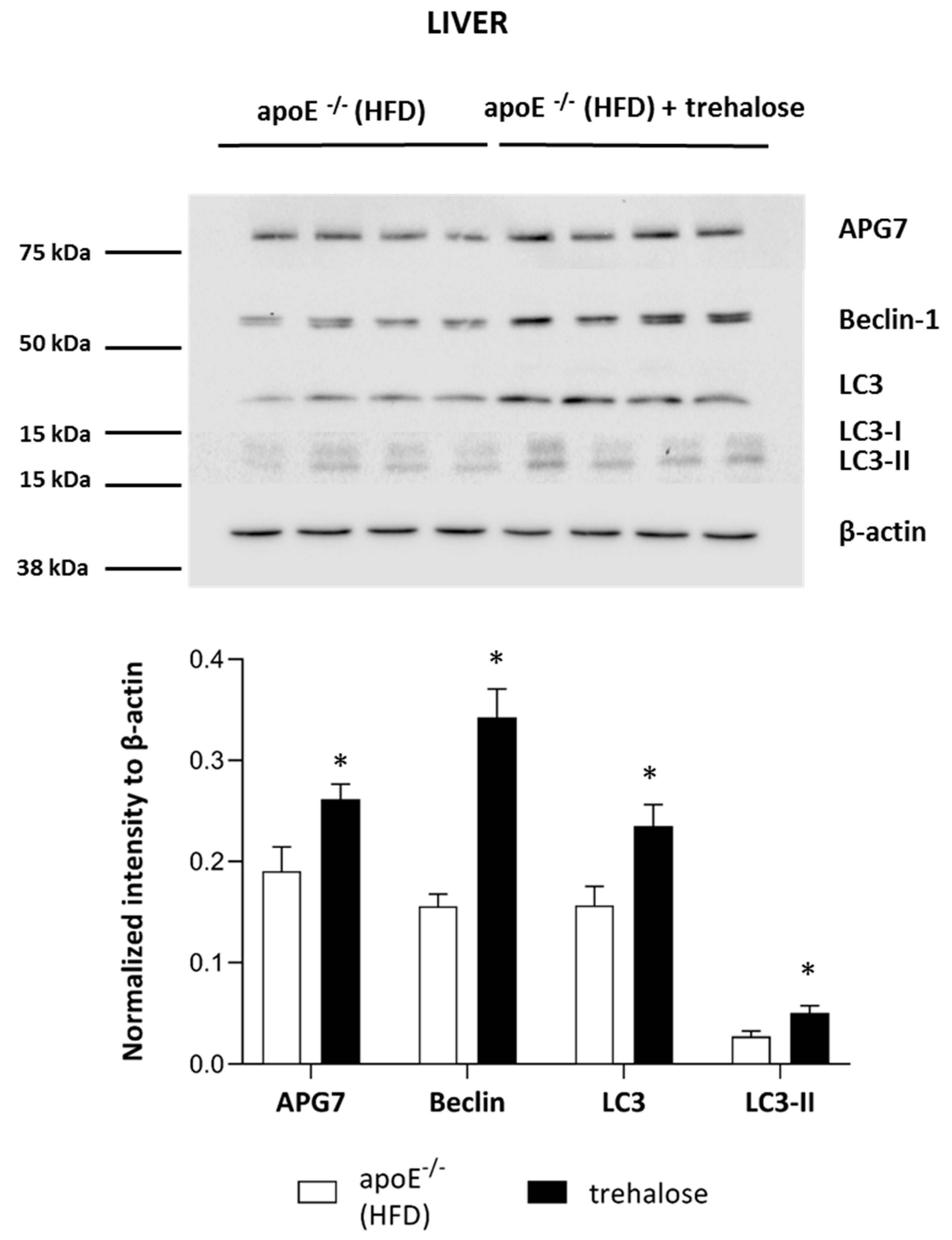
2.3. The Influence of Trehalose on Autophagy in The Liver
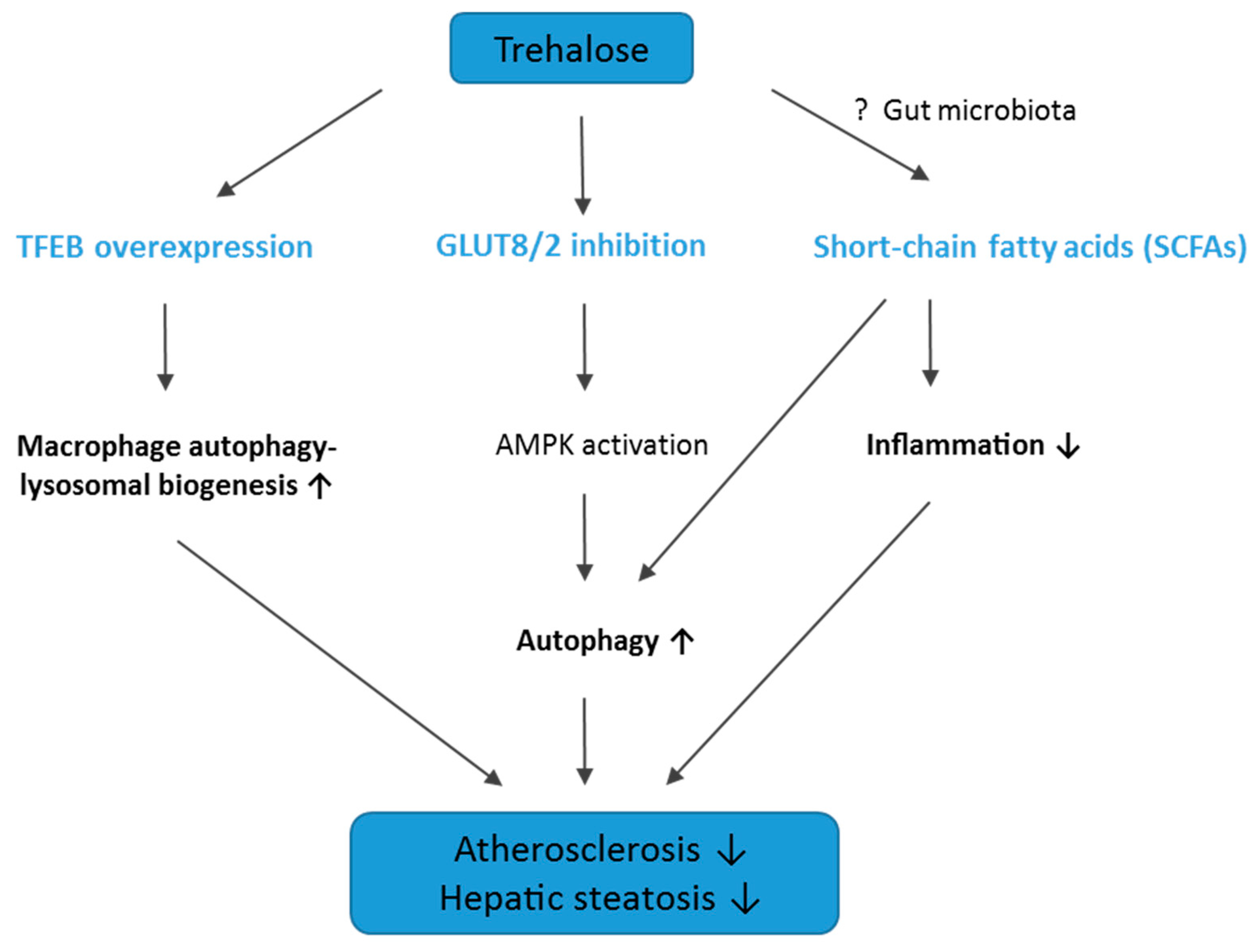
3. Discussion
3.1. The Influence of Trehalose on Atherosclerosis
3.2. The Influence of Trehalose on Hepatic Steatosis
3.3. Conclusions, Limitations of The Study, and Future Directions
4. Materials and Methods
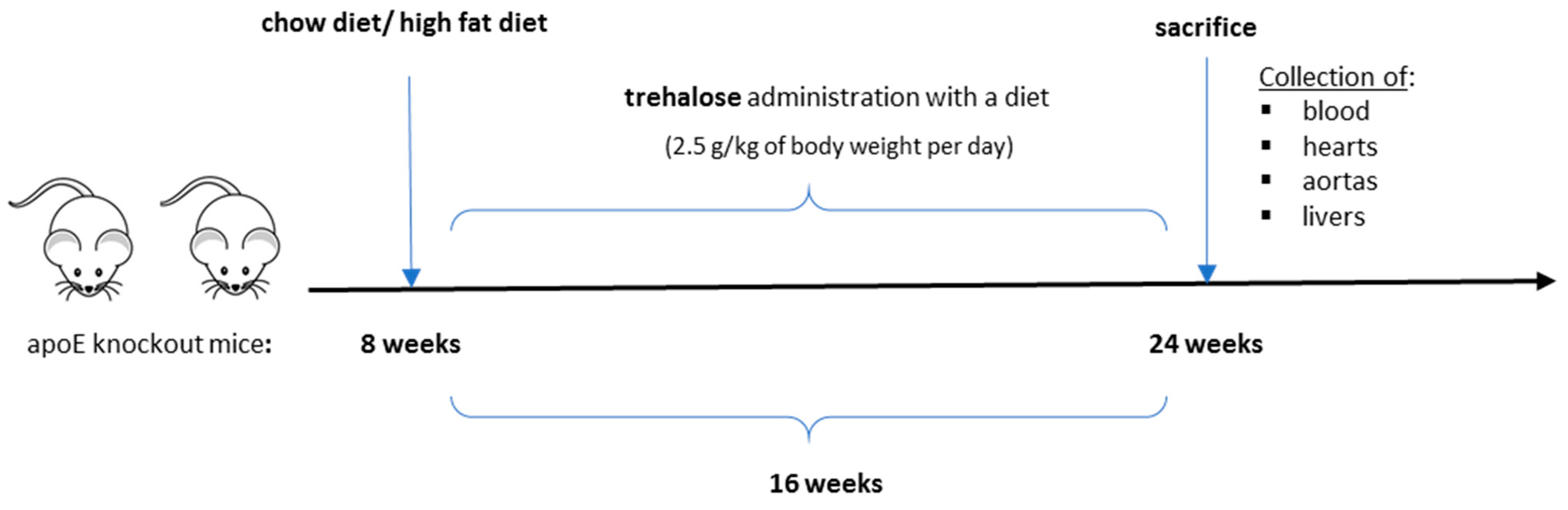
4.1. Animal Experiments
4.2. Atherosclerosis Studies
4.3. Immunohistochemistry of Aortic Roots
4.4. Histology of The Liver
4.5. Biochemical Methods
4.6. Western Blot
4.7. Real-Time PCR
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CD | chow diet |
| HE | hematoxylin/eosin |
| HFD | high-fat diet |
| NAFLD | nonalcoholic fatty liver disease |
References
- Lloyd-Jones, D.M. Cardiovascular risk prediction: Basic concepts, current status, and future directions. Circulation 2010, 121, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in atherosclerosis: Transition from theory to practice. Circ. J. 2010, 74, 213–220. [Google Scholar] [CrossRef]
- Targher, G. Non-alcoholic fatty liver disease, the metabolic syndrome and the risk of cardiovascular disease: The plot thickens. Diabet. Med. 2007, 24, 1–6. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Shao, B.; Han, B.; Zeng, Y.; Su, D.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Huda, N.; Khambu, B.; Yin, X.-M. Relevance of autophagy to fatty liver diseases and potential therapeutic applications. Amino Acids 2017, 49, 1965–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17R–27R. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.-M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Shukla, E.; Thorat, L.J.; Nath, B.B.; Gaikwad, S.M. Insect trehalase: Physiological significance and potential applications. Glycobiology 2015, 25, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Haddad, G.G. Role of trehalose phosphate synthase and trehalose during hypoxia: From flies to mammals. J. Exp. Biol. 2004, 207, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization; World Health Organization. Expert Committee on Food Additives Evaluation of Certain Food Additives and Contaminants; World Health Organization Technical Report Series; World Health Organization: Geneva, Switzerland, 2001. [Google Scholar]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal 2016, 9, ra21. [Google Scholar] [CrossRef] [PubMed]
- Arai, C.; Arai, N.; Mizote, A.; Kohno, K.; Iwaki, K.; Hanaya, T.; Arai, S.; Ushio, S.; Fukuda, S. Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance. Nutr. Res. 2010, 30, 840–848. [Google Scholar] [CrossRef]
- Davies, J.E.; Sarkar, S.; Rubinsztein, D.C. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2006, 15, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Hatamipour, M.; Tabatabaei, S.A. Trehalose administration attenuates atherosclerosis in rabbits fed a high-fat diet. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Stachowicz, A.; Suski, M.; Olszanecki, R.; Madej, J.; Okoń, K.; Korbut, R. Proteomic analysis of liver mitochondria of apolipoprotein E knockout mice treated with metformin. J Proteom. 2012, 77, 167–175. [Google Scholar] [CrossRef]
- Kolovou, G.; Anagnostopoulou, K.; Mikhailidis, D.P.; Cokkinos, D.V. Apolipoprotein E knockout models. Curr. Pharm. Des. 2008, 14, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Surra, J.C.; Guillén, N.; Arbonés-Mainar, J.M.; Barranquero, C.; Navarro, M.A.; Arnal, C.; Orman, I.; Segovia, J.C.; Osada, J. Sex as a profound modifier of atherosclerotic lesion development in apolipoprotein E-deficient mice with different genetic backgrounds. J. Atheroscler. Thromb. 2010, 17, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Maybüchen, L.; Zimmer, S.; Hittatiya, K.; Bäck, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, T.D.; Jeong, S.-J.; Zhang, X.; Sergin, I.; Razani, B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 2018, 14, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Autophagy in atherosclerosis: A potential drug target for plaque stabilization. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2787–2791. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef] [PubMed]
- Arai, C.; Miyake, M.; Matsumoto, Y.; Mizote, A.; Yoshizane, C.; Hanaya, Y.; Koide, K.; Yamada, M.; Hanaya, T.; Arai, S.; et al. Trehalose prevents adipocyte hypertrophy and mitigates insulin resistance in mice with established obesity. J. Nutr. Sci. Vitaminol. 2013, 59, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Pagliassotti, M.J.; Estrada, A.L.; Hudson, W.M.; Wei, Y.; Wang, D.; Seals, D.R.; Zigler, M.L.; LaRocca, T.J. Trehalose supplementation reduces hepatic endoplasmic reticulum stress and inflammatory signaling in old mice. J. Nutr. Biochem. 2017, 45, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Mizote, A.; Yamada, M.; Yoshizane, C.; Arai, N.; Maruta, K.; Arai, S.; Endo, S.; Ogawa, R.; Mitsuzumi, H.; Ariyasu, T.; et al. Daily Intake of Trehalose Is Effective in the Prevention of Lifestyle-Related Diseases in Individuals with Risk Factors for Metabolic Syndrome. J. Nutr. Sci. Vitaminol. 2016, 62, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, N. Glycosidase inhibitors: Update and perspectives on practical use. Glycobiology 2003, 13, 93R–104R. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.B.; Krakowka, S.; Dexter, L.B.; Schmid, H.; Wolterbeek, A.P.M.; Waalkens-Berendsen, D.H.; Shigoyuki, A.; Kurimoto, M. Trehalose: A review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem. Toxicol. 2002, 40, 871–898. [Google Scholar] [CrossRef]
- Bergoz, R.; Bolte, J.P. Meyer zum Bueschenfelde, null Trehalose tolerance test. Its value as a test for malabsorption. Scand. J. Gastroenterol. 1973, 8, 657–663. [Google Scholar]
- Iannucci, L.F.; Sun, J.; Singh, B.K.; Zhou, J.; Kaddai, V.A.; Lanni, A.; Yen, P.M.; Sinha, R.A. Short chain fatty acids induce UCP2-mediated autophagy in hepatic cells. Biochem. Biophys. Res. Commun. 2016, 480, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Pawlowska, M.; Gajda, M.; Pyka-Fosciak, G.; Toton-Zuranska, J.; Niepsuj, A.; Kus, K.; Bujak-Gizycka, B.; Suski, M.; Olszanecki, R.; Jawien, J.; et al. The effect of doxycycline on atherogenesis in apoE-knockout mice. J. Physiol. Pharmacol. 2011, 62, 247–250. [Google Scholar]
- Stachowicz, A.; Olszanecki, R.; Suski, M.; Wiśniewska, A.; Totoń-Żurańska, J.; Madej, J.; Jawień, J.; Białas, M.; Okoń, K.; Gajda, M.; et al. Mitochondrial aldehyde dehydrogenase activation by alda-1 inhibits atherosclerosis and attenuates hepatic steatosis in apolipoprotein e-knockout mice. J. Am. Heart Assoc. 2014, 3, e001329. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total Cholesterol [mmol/L] | HDL [mmol/L] | LDL [mmol/L] | TG [mmol/L] |
|---|---|---|---|---|
| apoE−/− (CD) | 13.4 ± 0.6 | 3.72 ± 0.95 | 8.24 ± 0.84 | 1.36 ± 0.22 |
| apoE−/− (CD) + trehalose | 12.0 ± 3.20 | 3.58 ± 0.30 | 7.4 ± 0.88 | 1.48 ± 0.57 |
| apoE−/− (HFD) | 27.24 ± 4.49 | 3.93 ± 0.83 | 22.51 ± 4.10 | 1.03 ± 0.22 |
| apoE−/− (HFD) + trehalose | 25.25 ± 4.77 | 5.06 ± 0.50 | 19.90 ± 4.49 | 0.67 ± 0.12 * |
| Components | Chow Diet (CD) | High Fat Diet (HFD) |
|---|---|---|
| Fat | 4% | 15.2% |
| Cholesterol | 0% | 0.25% |
| Protein | 17% | 16.9% |
| Fiber | 6.5% | 5.4% |
| Ash | 5.6% | 5.3% |
| Others | 66.9% | 57% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachowicz, A.; Wiśniewska, A.; Kuś, K.; Kiepura, A.; Gębska, A.; Gajda, M.; Białas, M.; Totoń-Żurańska, J.; Stachyra, K.; Suski, M.; et al. The Influence of Trehalose on Atherosclerosis and Hepatic Steatosis in Apolipoprotein E Knockout Mice. Int. J. Mol. Sci. 2019, 20, 1552. https://doi.org/10.3390/ijms20071552
Stachowicz A, Wiśniewska A, Kuś K, Kiepura A, Gębska A, Gajda M, Białas M, Totoń-Żurańska J, Stachyra K, Suski M, et al. The Influence of Trehalose on Atherosclerosis and Hepatic Steatosis in Apolipoprotein E Knockout Mice. International Journal of Molecular Sciences. 2019; 20(7):1552. https://doi.org/10.3390/ijms20071552
Chicago/Turabian StyleStachowicz, Aneta, Anna Wiśniewska, Katarzyna Kuś, Anna Kiepura, Anna Gębska, Mariusz Gajda, Magdalena Białas, Justyna Totoń-Żurańska, Kamila Stachyra, Maciej Suski, and et al. 2019. "The Influence of Trehalose on Atherosclerosis and Hepatic Steatosis in Apolipoprotein E Knockout Mice" International Journal of Molecular Sciences 20, no. 7: 1552. https://doi.org/10.3390/ijms20071552
APA StyleStachowicz, A., Wiśniewska, A., Kuś, K., Kiepura, A., Gębska, A., Gajda, M., Białas, M., Totoń-Żurańska, J., Stachyra, K., Suski, M., Jawień, J., Korbut, R., & Olszanecki, R. (2019). The Influence of Trehalose on Atherosclerosis and Hepatic Steatosis in Apolipoprotein E Knockout Mice. International Journal of Molecular Sciences, 20(7), 1552. https://doi.org/10.3390/ijms20071552