Bridging the Gap between Alzheimer’s Disease and Alzheimer’s-like Diseases in Animals



Abstract
:
1. Introduction
2. Aging and Pathogenesis of Alzheimer’s Disease (AD)
3. Inflammatory and Immune Responses to AD
4. Animals and Neurodegenerative Diseases Including Alzheimer’s-like Disease
5. Identification of the Disease
6. Treatment Strategies
7. Conclusions
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| ADAM | A disintegrin- and metalloproteinase-family enzymes |
| AICD | APP intracellular domain |
| ALD | Alzheimer’s–like disease |
| APP | Amyloid precursor protein |
| Aβ | Amyloid β |
| ROS | Reactive oxygen species |
| BACE1 | Beta-secretase 1 |
| BBB | Blood brain barrier |
| C83 | 83-amino acid C-terminal fragment |
| C99 | 99-amino acid C terminal fragment |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CTF83 | 83-amino acid C-terminal fragment |
| FDG-PET | Fluorodeoxyglucose positron emission tomography |
| GSK-3β | Glycogen synthase kinase-3 beta |
| HCSMA | Hereditary canine spinal muscular atrophy |
| MAP | Microtubule associated protein |
| MCI | Mild cognitive impairment |
| MHC | Major histocompatibility complex |
| MND | Motor neuron disease |
| MRI | Magnetic resonance imaging |
| NFT | Neurofibrillary tangles |
| PET | Positron emission tomography |
| RNS | Reactive nitrogen species |
| sAPPα | Soluble amyloid precursor protein α |
| sAPPβ | Soluble amyloid precursor protein β |
| SP | Senile plaques |
| SR | Scavenger receptors |
| T2D | Type 2 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TLR | Toll-like receptors |
| TPK | Tau protein kinase |
References
- Melis, J.P.M.; Jonker, M.J.; Vijg, J.; Hoeijmakers, J.H.J.; Breit, T.M.; Van Steeg, H. Aging on a different scale—Chronological versus pathology-related aging. Aging 2013, 10, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. Longevity and sexual maturity vary across species with number of cortical neurons, and humans are no exception. J. Comp. Neurol. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/cne.24564 (accessed on 30 November 2018). [CrossRef] [PubMed]
- WHO. Facts about Ageing. Available online: http://www.who.int/ageing/about/facts/en/ (accessed on 30 October 2018).
- American Veterinary Medical Association. U.S. Pet Ownership & Demographics Sourcebook; American Veterinary Medical Association: Schaumburg, IL, USA, 2012. [Google Scholar]
- Alzheimer’s Disease International. World Alzheimer Report 2018. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2018.pdf (accessed on 14 March 2019).
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef]
- WHO. Nomenclature of amyloid and amyloidosis. Bull. World Health Organ. 1993, 71, 105–108. [Google Scholar]
- Primakoff, P.; Myles, D.G. The ADAM gene family: Surface proteins with adhesion and protease activity. Trends Genet. 2000, 16, 83–87. [Google Scholar] [CrossRef]
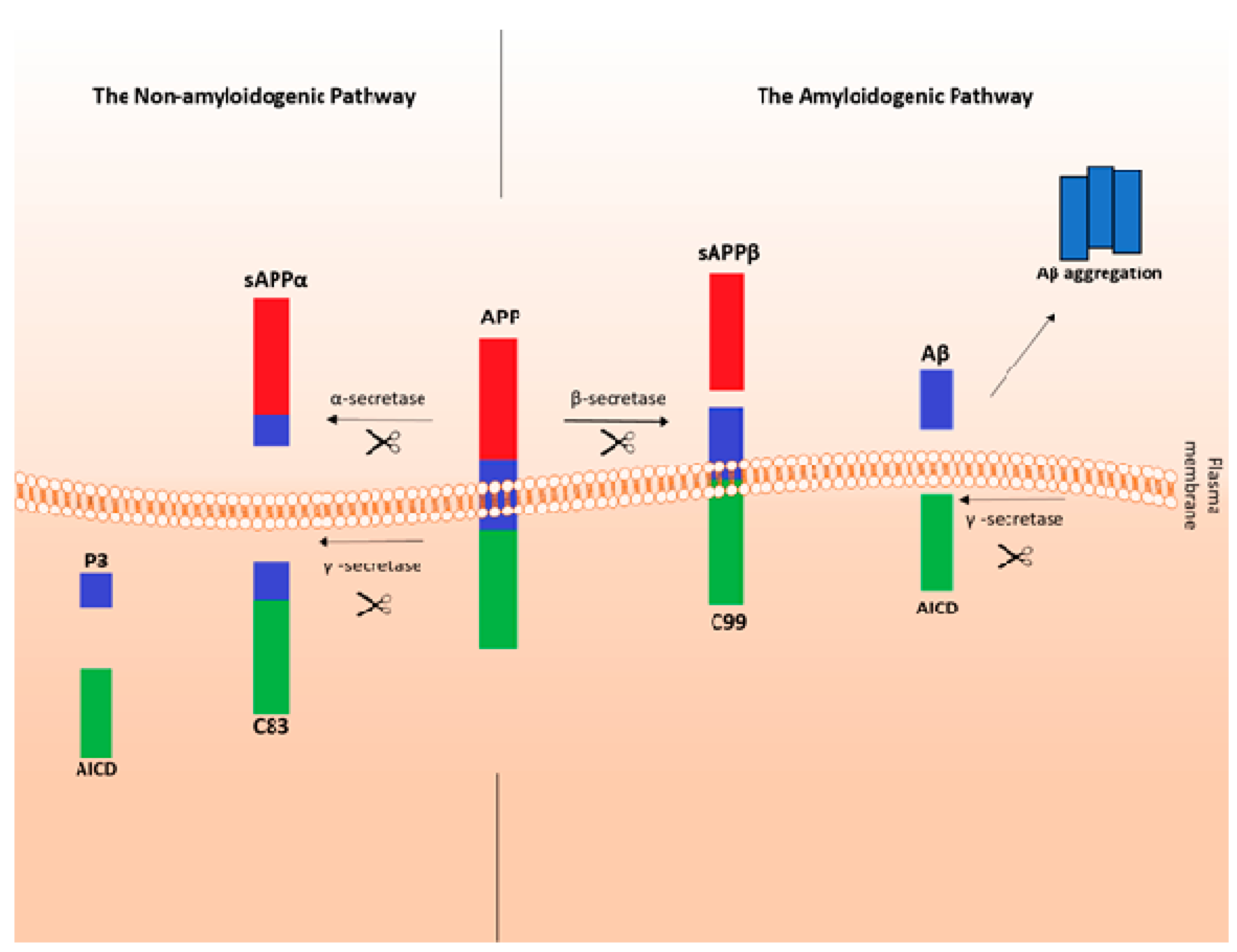
- Kojro, E.; Fahrenholtz, F. The non-amyloidogenic pathway: Structure and function of alpha-secretases. Subcell. Biochem. 2005, 38, 105–127. [Google Scholar]
- Plummer, S.; Van den Heuvel, C.; Thornton, E.; Corrigan, F.; Cappai, R. The Neuroprotective Properties of the Amyloid Precursor Protein Following Traumatic Brain Injury. Aging Dis. 2016, 7, 163–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, A.; Sawmiller, D.; Tan, J. Restoring sAPPα functions as a potential treatment for Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 973–991. [Google Scholar] [CrossRef] [PubMed]
- Youssef, S.A.; Capucchio, M.T.; Rofina, J.E. Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Vet. Pathol. 2016, 145, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimers Dis. 2010, 13, 311. [Google Scholar] [CrossRef]
- Soelke, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Kontush, A.; Berndt, C.; Weber, W.; Akopyan, V.; Arlt, S.; Beisiegel, U. Amyloid beta is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic. Biol. Med. 2001, 30, 119–128. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-Beta: A Crucial Factor in Alzheimer’s Disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Talebi, M.; Farhoudi, M. Beta-amyloid exhibits antagonistic effects on alpha 7 nicotonic acetylocholine receptors in orchestrated manner. Med. Hypotheses 2014, 8, 49–52. [Google Scholar] [CrossRef]
- McIntee, F.L.; Giannoni, P.; Blais, S.; Sommer, G.; Neubert, T.A.; Rostagno, A.; Ghiso, J. In vivo Differential Brain Clearance and Catabolism of Monomeric and Oligomeric Alzheimer’s Aβ protein. Front. Aging Neurosci. 2016, 27, 223. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Frasca, A.; Zotti, M.; La Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav. Immun. 2017, 60, 18–197. [Google Scholar] [CrossRef]
- Youssef, S.A. Pathology of Brain Aging and Animal Models of Neurodegenerative Diseases. In Conn’s Handbook of Models for Human Aging, 2nd ed.; Ram, J., Conn, P.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; Chapter 66; pp. 899–908. [Google Scholar]
- Kovacs, G.G. Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative Stress, Protein Modification and Alzheimer Disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Gold, M.; El Khoury, J. β-amyloid, microglia, and the inflammasome in Alzheimer’s disease. Semin. Immunopathol. 2015, 37, 607–611. [Google Scholar] [CrossRef]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef]
- Yan, Q.; Zhang, J.; Liu, H. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J. Neurosci. 2003, 23, 7504–7509. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Luster, A.D.; Terada, K.; Geula, C. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rube, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Gołąb, J.; Jakóbisiak, M.; Lasek, W.; Stokłosa, T. Prezentacja antygenów w ośrodkowym układzie nerwowym w kontekście bariery krew–mózg. In Immunologia, 5th ed.; Wydawnictwo Naukowe PWN: Warsaw, Poland, 2010; pp. 300–302. [Google Scholar]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Cappelano, G.; Careccchio, M.; Fleetwood, T.; Magistrelli, L.; Cantello, R.; Dianzani, U.; Comi, C. Immunity and Inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 89–107. [Google Scholar]
- Comi, C.; Tondo, G. Insights into the protective role of immunity in neurodegenerative disease. Neural Regen. Res. 2017, 12, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Sainaghi, P.P.; Bellan, M.; Lombino, F.; Alciato, F.; Carecchio, M.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; Cantello, R.; Pirisi, M.; et al. Growth Arrest Specific 6 Concentration is Increased in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2016, 55, 59–65. [Google Scholar] [CrossRef]
- Ota, Y.; Zanetti, A.T.; Hallock, R.M. The role of astrocytes in the regulation of synaptic plasticity and memory formation. Neural Plast. 2013. Available online: https://www.hindawi.com/journals/np/2013/185463/cta/ (accessed on 31 October 2018). [CrossRef] [PubMed]
- Yu, Y.; Ye, R.D. Microglial Aβ Receptors in Alzheimer’s Disease. Cell Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef]
- Su, F.; Bai, F.; Zhou, H.; Zhang, Z. Reprint of: Microglial toll-like receptors and Alzheimer’s disease. Brain Behav. Immun. 2016, 55, 166–178. [Google Scholar] [CrossRef]
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Ab uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, A.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: Amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schaffer, W.; et al. Role of the Toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Letiembre, M.; Liu, Y.; Walter, S.; Hao, W.; Pfander, T.; Wrede, A.; Schulz-Schaeffer, W.; Fassbender, K. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol. Aging 2009, 30, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Akiyama, H.; Yamada, Y.; Kondo, H.; Kawabe, Y.; Takeya, M.; Takahashi, K.; Suzuki, H.; Doi, T.; Sakamoto, A.; et al. Immunohistochemical evidence for a macrophage scavenger receptor in Mato cells and reactive microglia of ischemia and Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1998, 245, 734–740. [Google Scholar] [CrossRef]
- Christie, R.H.; Freeman, M.; Hyman, B.T. Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer’s disease. Am. J. Pathol. 1996, 148, 399–403. [Google Scholar]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef]
- Husemann, J.; Loike, J.D.; Kodama, T.; Silverstein, S.C. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J. Neuroimmunol. 2001, 114, 142–150. [Google Scholar] [CrossRef]
- Frenkel, D.; Wilkinson, K.; Zhao, L.; Hickman, S.E.; Means, T.K.; Puckett, L.; Farfara, D.; Kingery, N.D.; Weiner, H.L.; El Khoury, J. Scara1 deficiency impairs clearance of soluble amyloid-b by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 2013, 4, 2030. [Google Scholar] [CrossRef]
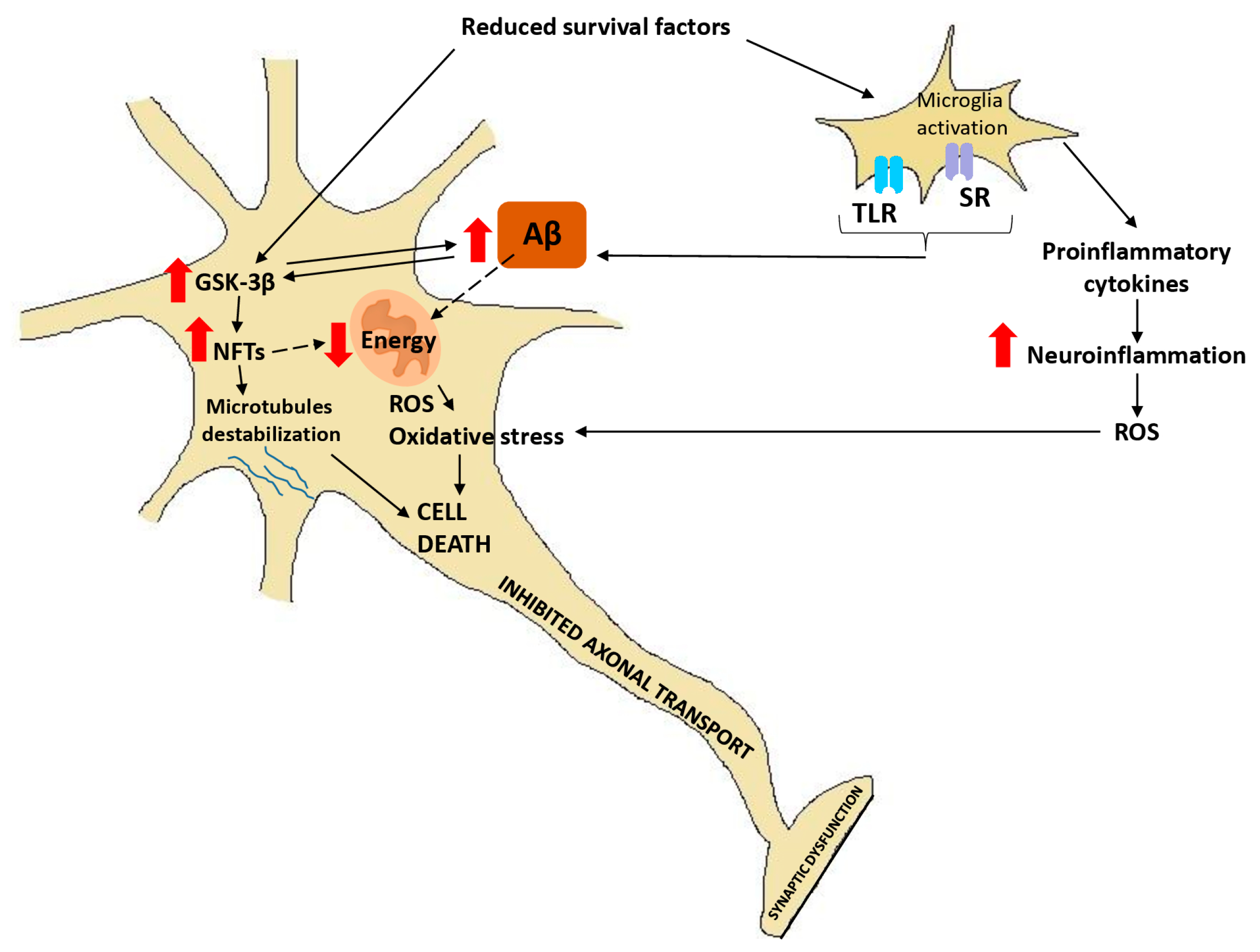
- Terwel, D.; Muyllaret, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Noguchi, K.; Michel, G.; Mercken, M.; Hoshi, M.; Ishiguro, K. Exposure of rat hippocampal neurons to amyloid beta peptide (25–35) induces the inactivation of phosphatidylinositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 1996, 203, 33–36. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Noh, M.Y.; Kim, S.H. Amyloid-beta-induced neurotoxicity is reduced by inhibition of glycogen synthase kinase-3. Brain Res. 2008, 1188, 254–262. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Rockenstein, E.; Torrance, M.; Adame, A.; Mante, M.; Bar-On, P.; Rose, J.B. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 2007, 27, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Neuronal plasticity and stressor toxicity during aging. Exp. Gerontol. 2000, 35, 1165–1183. [Google Scholar] [CrossRef]
- Teter, B.; Ashford, J.W. Neuroplasticity in Alzheimer’s Disease. J. Neurosci. Res. 2002, 70, 402–437. [Google Scholar] [CrossRef] [PubMed]
- Tsartsalis, S.; Xekardaki, A.; Hof, P.R.; Kovari, E.; Bouras, C. Early Alzheimer-type lesions in cognitively normal subjects. Neurobiol. Aging 2018, 62, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; Melo-Pires, M.; Sousa, L.; Sousa, N. Does the Interplay Between Aging and Neuroinflammation Modulate Alzheimer’s Disease Clinical Phenotypes? A Clinico-Pathological Perspective. J. Alzheimers Dis. 2016, 53, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Stockard, C.R. A hereditary lethal for localized motor and preganglionic neurones with a resulting paralysis in the dog. Am. J. Anat. 1936, 59, 1–53. [Google Scholar] [CrossRef]
- Cummings, J.F.; de Lahunta, A. A study of cerebellar and cerebral cortical degeneration in miniature poodle pups with emphasis on the ultrastructure of Purkinje cell changes. Acta Neuropathol. 1988, 75, 61–71. [Google Scholar] [CrossRef]
- Siso, S.; Pumarola, M.; Ferrer, I. Cell death and decreased synaptic protein expression in the ventral horn of Holstein-Friesian calves with spinal muscular atrophy. J. Comp. Pathol. 2003, 128, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.L.; Gilmore, W.C.; Litke, L.L. Motor neuron disease with neurofilamentous accumulations in Hampshire pigs. J. Vet. Diagn Investig. 1989, 1, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Pumarola, M.; Fondevila, D.; Majo, N.; Borras, D.; Ferrer, I. Neuronal vacuolation in young Rottweiler dogs. Acta Neuropathol. 1999, 97, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Siso, S.; Hanzlicek, D.; Fluehmann, G.; Kathmann, I.; Tomek, A.; Papa, V.; Vandevelde, M. Neurodegenerative diseases in domestic animals: A comparative review. Vet. J. 2006, 171, 20–38. [Google Scholar] [CrossRef]
- Landsberg, G.M.; Ruehl, W.W. Geriatric behavioral problems. J. Small Anim. Pract. 1997, 27, 1537–1559. [Google Scholar] [CrossRef]
- Houpt, K.A. Cognitive dysfunction in geriatric cats. In Consultations in Feline Internal Medicine, 4th ed.; August, J.R., Ed.; WB Saunders: Philadelphia, PA, USA, 2001; pp. 583–859. [Google Scholar]
- Landsberg, G.L.; Hunthausen, W.; Ackerman, L. The effects of aging on behavior in senior pets. In Handbook of Behavior Problems in the Dog and Cat, 2nd ed.; Landsberg, G.M., Hunthausen, W., Ackerman, L., Saunders, W.B., Eds.; WB Saunders: Philadelphia, PA, USA, 2003; pp. 269–304. [Google Scholar]
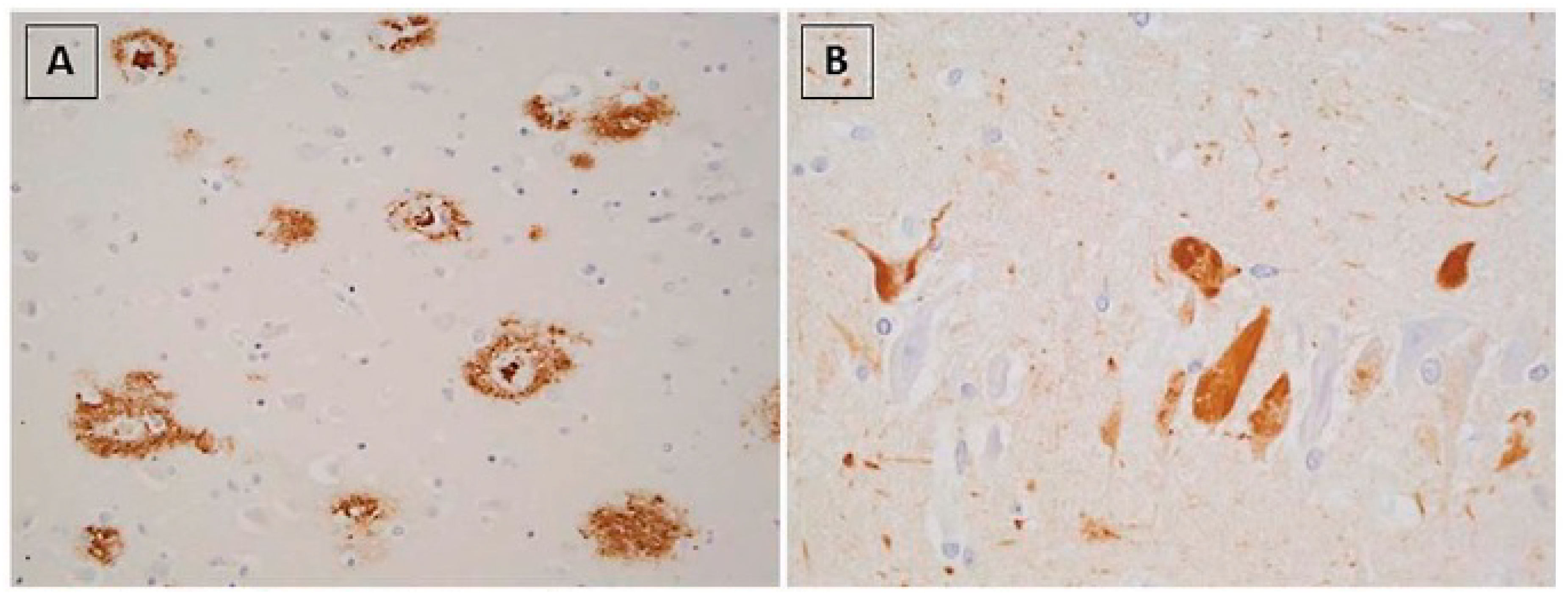
- Brellou, G.; Vlemmas, I.; Lekkas, S.; Papaioannou, N. Immunohistochemical investigation of amyloid beta-protein (Abeta) in the brain of aged cats. Histol. Histopathol. 2005, 20, 725–731. [Google Scholar]
- Head, E.; Moffat, K.; Das, P.; Sarsoza, F.; Poon, W.W.; Landsberg, G.; Cotman, C.W.; Murphy, M.P. Beta-Amyloid deposition and tau phosphorylation in clinically characterized aged cats. Neurobiol. Aging 2005, 26, 749–763. [Google Scholar] [CrossRef]
- Chambers, J.K.; Tokuda, T.; Uchida, K.; Ishii, R.; Tatebe, H.; Takahashi, E.; Tomiyama, T.; Une, Y.; Nakayama, H. The domestic cat as a natural animal model of Alzheimer’s disease. Acta Neuropathol. Commun. 2015, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.J.; Head, E.; Milgram, N.W.; Cotman, C.W. The canine as an animal model of human aging and dementia. Neurobiol. Aging 1996, 17, 259–268. [Google Scholar] [CrossRef]
- Gunn-Moore, D.; Moffat, K.; Christie, L.A.; Head, E. Cognitive dysfunction and the neurobiology of ageing in cats. J. Small Anim. Pract. 2007, 48, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Szabo, D.; Gee, N.R.; Miklosi, A. Natural or pathologic? Discrepancies in the study of behavioural and cognitive signs in aging family dogs. J. Vet. Behav. 2016, 11, 86–98. [Google Scholar] [CrossRef]
- Schütt, T.; Helboe, L.; Pedersen, L.O.; Waldemar, G.; Mette, B.; Pedersen, J.T. Dogs with Cognitive Dysfunction as a Spontaneous Model for Early Alzheimer’s Disease: A Translational Study of Neuropathological and Inflammatory Markers. J. Alzheimers Dis. 2016, 52, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Salvin, H.E.; McGreevy, P.D.; Sachdev, P.S.; Valenzuela, M.J. The canine cognitive dysfunction rating scale (CCDR): A data-driven and ecologically relevant assessment tool. Vet. J. 2011, 188, 331–336. [Google Scholar] [CrossRef]
- Rofina, J.E.; Van Ederen, A.M.; Toussaint, M.J.M.; Secreve, M.; Van der Spek, A.; Van der Meer, I.; Van Eerdenburg, F.J.; Gruys, E. Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res. 2006, 1069, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Su, M.Y.; Tapp, P.D.; Vu, L.; Chen, Y.F.; Chu, Y.; Muggenburg, B.; Chiou, J.Y.; Chen, C.; Wang, J.; Bracco, C.; et al. A longitudinal study of brain morphometrics using serial magnetic resonance imaging analysis in a canine model of aging. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Head, E.; Pop, V.; Sarsoza, F.; Kayed, R.; Beckett, T.L.; Studzinski, C.M.; Tomic, J.L.; Glabe, C.G.; Murphy, M.P. Amyloid-beta peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J. Alzheimers Dis. 2010, 20, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Insua, D.; Corredoira, A.; Gonzalez-Martinez, A.; Suarez, M.L.; Santamarina, G.; Sarasa, M.; Pesini, P. Expression of p75(NTR), a marker for basal forebrain cholinergic neurons, in young and aged dogs with or without cognitive dysfunction syndrome. J. Alzheimers Dis. 2012, 28, 291–296. [Google Scholar] [CrossRef]
- Head, E. A canine model of human aging and Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1832, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Borras, D.; Ferrer, I.; Pumarola, M. Age-related Changes in the Brain of the Dog. Vet. Pathol. 1999, 36, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Russell, M.J.; White, R.; Patel, E.; Markesbery, W.R.; Watson, C.R.; Geddes, J.W. Familial influence on plaque formation in the beagle brain. Neuroreport 1992, 12, 1093–1096. [Google Scholar] [CrossRef]
- Wegiel, J.; Wisniewski, H.M.; Dziewiatkowski, J.; Tarnawski, M.; Dziewiatkowska, A.; Morys, J.; Soltysiak, Z.; Kim, K.S. Subpopulation of dogs with severe brain parenchymal beta amyloidosis distinguished with cluster analysis. Brain Res. 1996, 728, 20–26. [Google Scholar] [CrossRef]
- Insua, D.; Gonzalez-Martinez, A.; Suarez, M.L.; Santamarina, G.; Sarasa, M.; Pesini, P. Dogs with canine counterpart of Alzheimer’s disease lose noradrenergic neurons. Neurobiol. Aging 2010, 31, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Capucchio, M.T.; Marquez, M.; Pregel, P.; Foradada, P.; Bravo, M.; Mattutino, G.; Torre, C.; Schiffer, D.; Catalano, D.; Valenza, F.; et al. Parenchymal and Vascular Lesions in Ageing Equine Brains Histological and Immunohistochemical Studies. J. Comp. Pathol. 2009, 142, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Greenberg, S.G.; Saper, C.B. Neurofibrillary tangles in the cerebral cortex of sheep. Neurosci. Lett. 1994, 170, 187–190. [Google Scholar] [CrossRef]
- Costassa, E.V.; Fiorini, M.; Zanusso, G.; Peletto, S.; Acutis, P.; Baioni, E.; Maurella, C.; Tagliavini, F.; Catania, M.; Gallo, M.; et al. Characterization of Amyloid-beta Deposits in Bovine Brains. J. Alzheimers Dis. 2016, 51, 875–887. [Google Scholar] [CrossRef]
- Cole, G.; Neal, J.W. The brain in aged elephants. J. Neuropathol. Exp. Neurol. 1990, 49, 190–192. [Google Scholar] [CrossRef]
- Nakamura, S.; Nakayama, H.; Uetsuka, K.; Sasaki, N.; Uchida, K.; Goto, N. Senile plaques in an aged two-humped (Bactrian) camel (Camelus bactrianus). Acta Neuropathol. 1995, 90, 415–418. [Google Scholar] [CrossRef]
- Gunn-Moore, D.; Kaidanovich-Beilin, O.; Iradi, M.C.G.; Gunn-Moore, F.; Lovestone, S. Alzheimer’s disease in humans and other animals: A consequence of postreproductive life span and longevity rather than aging. Alzheimers Dement. 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Ndung’u, M.; Härtig, W.; Wegner, F.; Mwenda, J.M.; Low, R.W.; Akinyemi, R.O.; Kalaria, R.N. Cerebral aβ (42) deposits and microvascular pathology in ageing baboons. Neuropathol. Appl. Neurobiol. 2011, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Masters, C.; Beyreuther, K.; Price, D.L. Amyloid in the brains of aged squirrel monkeys. Acta Neuropathol. 1990, 80, 381–387. [Google Scholar] [CrossRef]
- Kimura, N.; Nakamura, S.; Goto, N.; Narushima, E.; Hara, I.; Shichiri, S.; Saitou, K.; Nose, M.; Hayashi, T.; Kawamura, S.; et al. Senile plaques in an aged lowland Western Gorilla. Exp. Anim. 2001, 50, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Gearing, M.; Rebeck, G.W.; Hyman, B.T.; Tigges, J.; Mirra, S.S. Neuropathology and apolipoprotein E profile of aged chimpanzees: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9382–9386. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.E.; Sherwood, C.C.; Cranfield, M.R.; Erwin, J.M.; Mudakikwa, A.; Hof, P.R.; Mufson, E.J. Early Alzheimer’s disease-type pathology in the frontal cortex of wild mountain gorillas (Gorilla beringei beringei). Neurobiol. Aging 2016, 39, 195–201. [Google Scholar] [CrossRef]
- Schulz, C.; Braak, E.; Tredici, K.; Hubbard, G.; Braak, H. Tau Pathology in Neurons and Glial Cells of Aged Baboons. Adv. Exp. Med. Biol. 2001, 487, 59–69. [Google Scholar]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Mielke, M.M.; Rissman, R.A.; Lista, S.; Vanderstichele, H.; Zetterberg, H.; Lewczuk, P.; Posner, H.; Hall, J.; Johnson, L.; et al. Blood-based biomarkers in Alzheimer disease: Current state of the science and a novel collaborative paradigm for advancing from discovery to clinic. Alzheimers Dement. 2017, 13, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, J.; Andreasson, U.; Pannee, J.; Bjerke, M.; Portelius, E.; Leinenbach, A.; Bittner, T.; Korecka, M.; Jenkins, R.G.; Vanderstichele, H.; et al. CSF Aβ1–42 – an excellent but complicated Alzheimer’s biomarker—A route to standardisation. Clin. Chim. Acta 2017, 467, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx 2004, 1, 213–225. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003, 2, 605–613. [Google Scholar] [CrossRef]
- Lewczuk, P.; Kamrowski-Kruck, H.; Peters, O.; Heuser, I.; Jessen, F.; Popp, J.; Burger, K.; Hampel, H.; Frolich, L.; Wolf, S. Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer’s disease: A multicenter study. Mol. Psychiatry 2010, 15, 138–145. [Google Scholar] [CrossRef]
- Zetterberg, H.; Andreasson, U.; Hansson, O.; Wu, G.; Sankaranarayanan, S.; Andersson, M.E.; Buchhave, P.; Londos, E.; Umek, R.M.; Minthon, L. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch. Neurol. 2008, 65, 1102–1107. [Google Scholar] [CrossRef]
- Zhong, Z.; Ewers, M.; Teipel, S.; Burger, K.; Wallin, A.; Blennow, K.; He, P.; McAllister, C.; Hampel, H.; Shen, Y. Levels of b-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch. Gen. Psychiatry 2007, 64, 718–726. [Google Scholar] [CrossRef]
- Portelius, E.; Price, E.; Brinkmalm, G.; Stiteler, M.; Olsson, M.; Persson, R. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging 2009, 32, 1090–1098. [Google Scholar]
- Seo, E.H.; Park, W.Y.; Choo, I.H. Structural MRI and Amyloid PET Imaging for Prediction of Conversion to Alzheimer’s Disease in Patients with Mild Cognitive Impairment: A Meta-Analysis. Psychiatry Investig. 2017, 14, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Yu, J.T.; Tan, L. Biomarkers for Preclinical Alzheimer’s Disease. J. Alzheimers Dis. 2014, 42, 1051–1069. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, S.S.; Nygaard, A.B.; Christensen, T. miRNA expression profiles in cerebrospinal fluid and blood of patients with Alzheimer’s disease and other types of dementia – an exploratory study. Transl. Neurodegener. 2016, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.F.; Wu, N.; Wang, L.; Li, J. Circulating MicroRNAs: A Novel Class of Potential Biomarkers for Diagnosing and Prognosing Central Nervous System Diseases. Cell Mol. Biol. 2013, 33, 601–613. [Google Scholar] [CrossRef]
- Iranifar, E.; Serehst, B.M.; Momeni, F.; Fadaei, E.; Mehr, M.H.; Ebrahimi, Z.; Rahmati, M.; Kharazinejad, E.; Mirzaei, H. Exosomes and microRNAs: New potential therapeutic candidates in Alzheimer disease therapy. J. Cell Physiol. 2018, 234, 2296–2305. [Google Scholar] [CrossRef]
- Hajipour, M.J.; Santoso, M.R.; Rezaee, F.; Aghaverdi, H.; Mahmoudi, M.; Perry, G. Advances in Alzheimer’s diagnosis and Therapy: The Implications of Nanotechnology. Trends Biotechnol. 2018, 35, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Gabin, J.M.; Tambs, K.; Saltvedt, I.; Sund, E.; Holmen, J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: The HUNT Study. Alzheimers Res. Ther. 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment Strategies Targeting Amyloid Beta-Protein. Cold Spring Harb. Perspect. Med. 2018, 9, a006387. [Google Scholar]
- Neumann, U.; Ufer, M.; Jacobson, L.H.; Rouzade-Dominguez, M.L.; Hueldal, G.; Kolly, C.; Luond, R.M.; Machauer, R.; Veenstra, S.J.; Hurth, K.; et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 2018, 11, e9316. [Google Scholar] [CrossRef]
- Studzinski, C.M.; MacKay, W.A.; Beckett, T.L.; Henderson, S.T.; Murphy, M.P.; Sullivan, P.G.; Burnham, W.M. Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-β precursor protein (APP) levels in the aged dog. Brain Res. 2008, 1226, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361, 6406. [Google Scholar] [CrossRef]
- Kivipelto, M.; Hakansson, K. A rare success against Alzheimer’s. Sci. Am. 2017, 316, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.C.; Campos, G.A.A.; Galante, P.R.; Biolchi, A.M.; Goncalves, J.C.; Lopes, K.S.; Mortari, M.R. Peptides isolated from animal venom as a platform for new therapeutics for the treatment of Alzheimer’s disease. Neuropeptides 2018, 67, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.H.; Lu, P.J.; Su, J.C. Two types of Russell’s viper revealed by variation in phospholipases A2 from venom of the subspecies. Toxicon 1999, 34, 99–109. [Google Scholar] [CrossRef]
- Wang, T.; Wang, S.W.; Zhang, Y.; Wu, X.F.; Peng, Y.; Cao, Z.; Ge, B.Y.; Wang, X.; Wu, Q.; Lin, J.T.; et al. Scorpion Venom Heat-Resistant Peptide (SVHRP) Enhances Neurogenesis and Neurite Outgrowth of Immature Neurons in Adult Mice by Up-Regulating Brain-Derived Neurotrophic Factor (BDNF). PLoS ONE 2014, 9, e109977. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ithurralde, D.; Silveira, R.; Barbeito, L.; Dajas, F. Fasciculin, a powerful anticholinesterase polypeptide from Dendroaspis angusticeps venom. Neurochem. Int. 1983, 5, 267–274. [Google Scholar] [CrossRef]
- Samson, A.; Scherf, T.; Eisenstein, M.; Chill, J.; Anglister, J. The mechanism for acetylcholine receptor inhibition by alpha-neurotoxins and species-specific resistance to alpha-bungarotoxin revealed by NMR. Neuron 2002, 35, 319–332. [Google Scholar] [CrossRef]
- Bosch, M.N.; Pugliese, M.; Andrade, C.; Gimeno-Bayon, J.; Mahy, N.; Rodriguez, M.J. Amyloid-β Immunotherapy Reduces Amyloid Plaques and Astroglial Reaction in Aged Domestic Dogs. Neurodegener. Dis. 2015, 15, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Creagan, E.T.; Bauer, B.A.; Thomley, B.S.; Borg, J.M. Animal-assisted therapy at Mayo Clinic: The time is now. Complement. Ther. Clin. Pract. 2015, 21, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Katare, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. 2016, 10S, s144–s149. [Google Scholar] [CrossRef]
- Piasecka-Markowicz, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A Future Therapy for Neurodegenerative Diseases. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar]
- Rotermund, C.; Machentanz, G.; Fitzergald, J. The therapeutic Potential of Metformin in Neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Darwish, M.; Hishikawa, N.; Yamashita, T.; Sato, K.; Takemoto, M.; Abe, K. Therapeutic effects of drug switching between acetylcholinesterase inhibitors in patients with Alzheimer’s disease. Geriatr. Gerontol. Int. 2017, 17, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, D.; Chan, P.; Bradley, J.; Lanctot, K.; Herrmann, N. Acetylcholinesterase inhibitors for treating dementia symptoms—A safety evaluation. Expert Opin. Drug Saf. 2017, 16, 1009–1019. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Species | Symptoms | Literature | |
|---|---|---|---|---|
| Neuropathological | Behavioral | |||
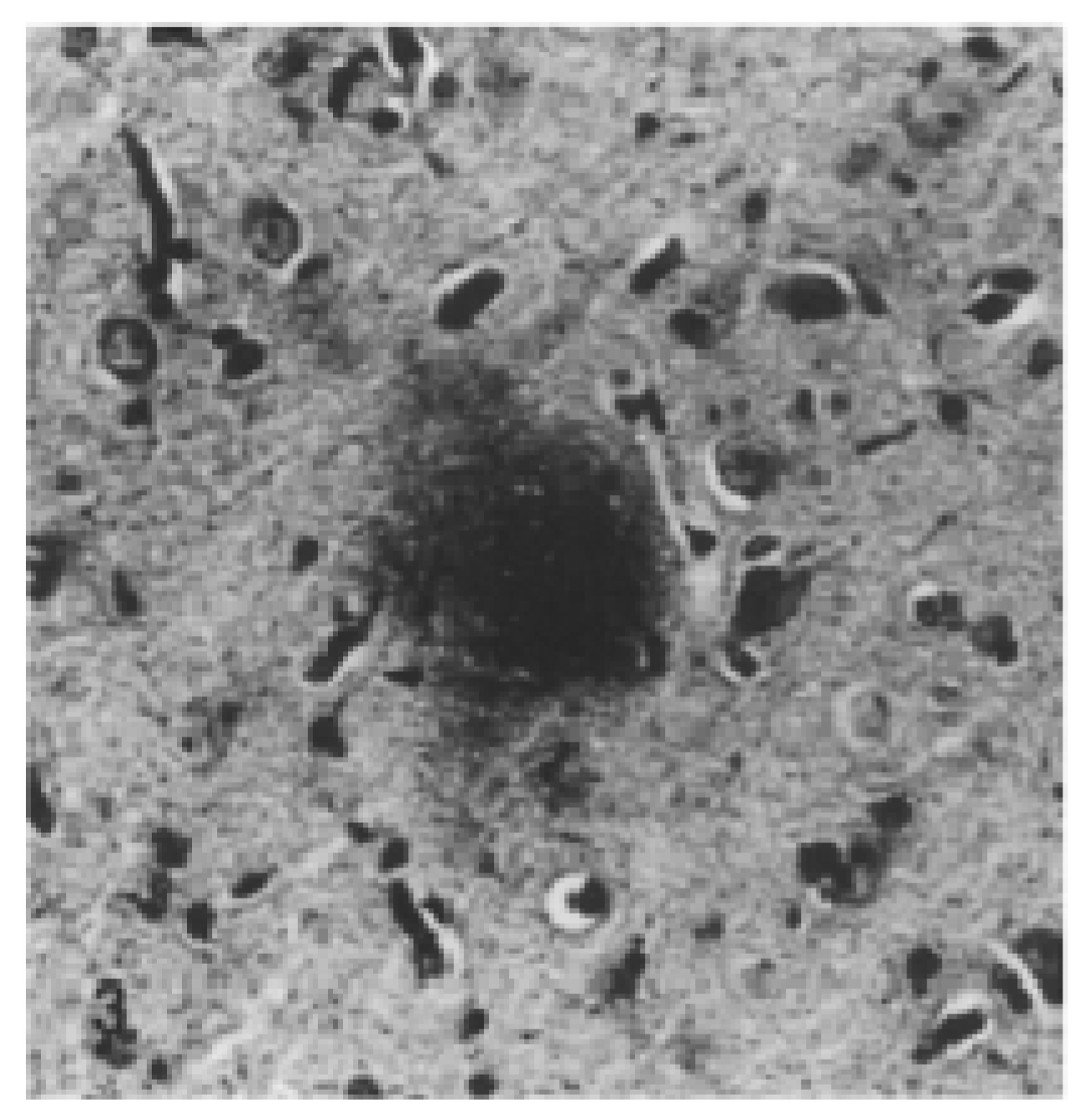
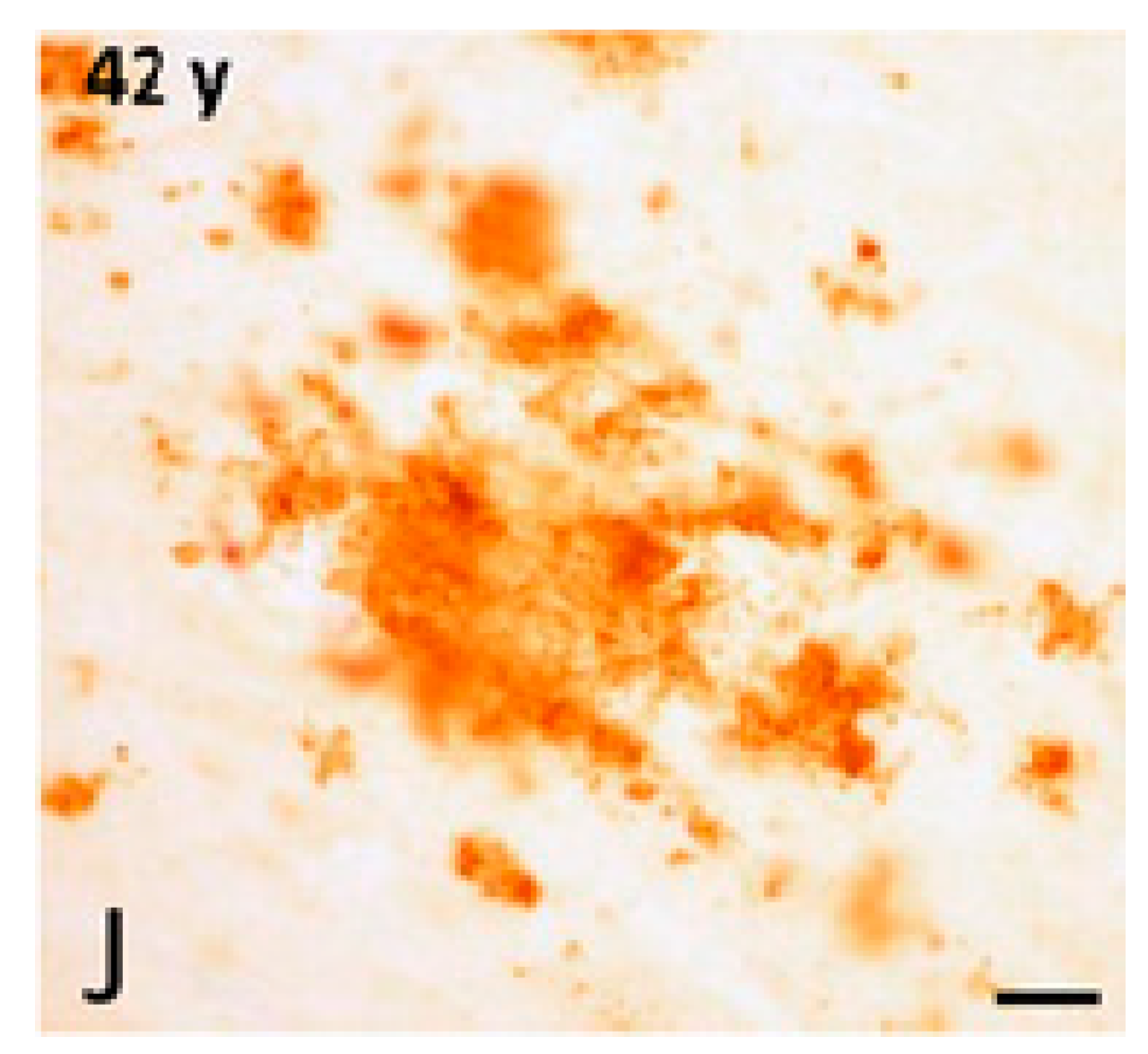
| Alzheimer’s disease | Human | Oxidative stress Neuroinflammation Disruption of neuronal cytoskeleton Impaired neuronal transport Synaptic dysfunction Senile plaques and Aβ aggregates NFTs, hyperphosphorylated tau accumulations Brain atrophy Neuronal loss | Loss of cognition and recognition Disorientation Apathy Poor judgment Intensified aggression or passiveness Changes in interest in food Poor judgment Difficulties in speaking, swallowing and walking Worsening memory | [18,19,21,22,23,24,25,56] |
| Alzheimer’s-like disease | Domestic cat | Aβ oligomers aggregates in cats aged 8 and more Senile plaques in cats aged 10 and more NFTs only in the presence of Aβ Neuronal loss Neuronal degradation | Spatial disorientation or confusion Altered social relationships Intensified aggression or passiveness Changes in daily schedule and wake-sleep pattern Changes in interest in food Decreased grooming Inappropriate vocalization | [66,67,68,69,70,71] |
| Camel | Diffuse-type senile plaques in the cerebral cortex | No data | [91] | |
| Elephant | Aβ cerebral deposition | [90] | ||
| Cattle | Aβ cerebral deposition | [89] | ||
| Horse | Tau accumulations Axonal transport deficiencies Diffuse Aβ plaques | [87] | ||
| Sea lion | CSF markers which are found in AD | [92] | ||
| Dolphin | Amyloid and tau pathology in the brain | [92] | ||
| Gorilla | Diffuse-type senile plaques Vascular amyloid | [95,97] | ||
| Baboon | Diffuse-type senile plaques Vascular amyloid Hyperphosphorylated tau in the oldest individuals | [93,98] | ||
| Squirrel monkey | Senile plaques Diffuse-type senile plaques Vascular amyloid | [94] | ||
| Chimpanzee | Diffuse-type senile plaques Vascular amyloid | [96] | ||
| Macaque | Senile plaques Vascular amyloid | [95] | ||
| Sheep | NFTs Aβ cerebral deposition | [88] | ||
| Cheetah | Amyloid plaques NFTs | [20] | ||
| Bears | Amyloid plaques NFTs | [20] | ||
| Goats | NFTs | [20] | ||
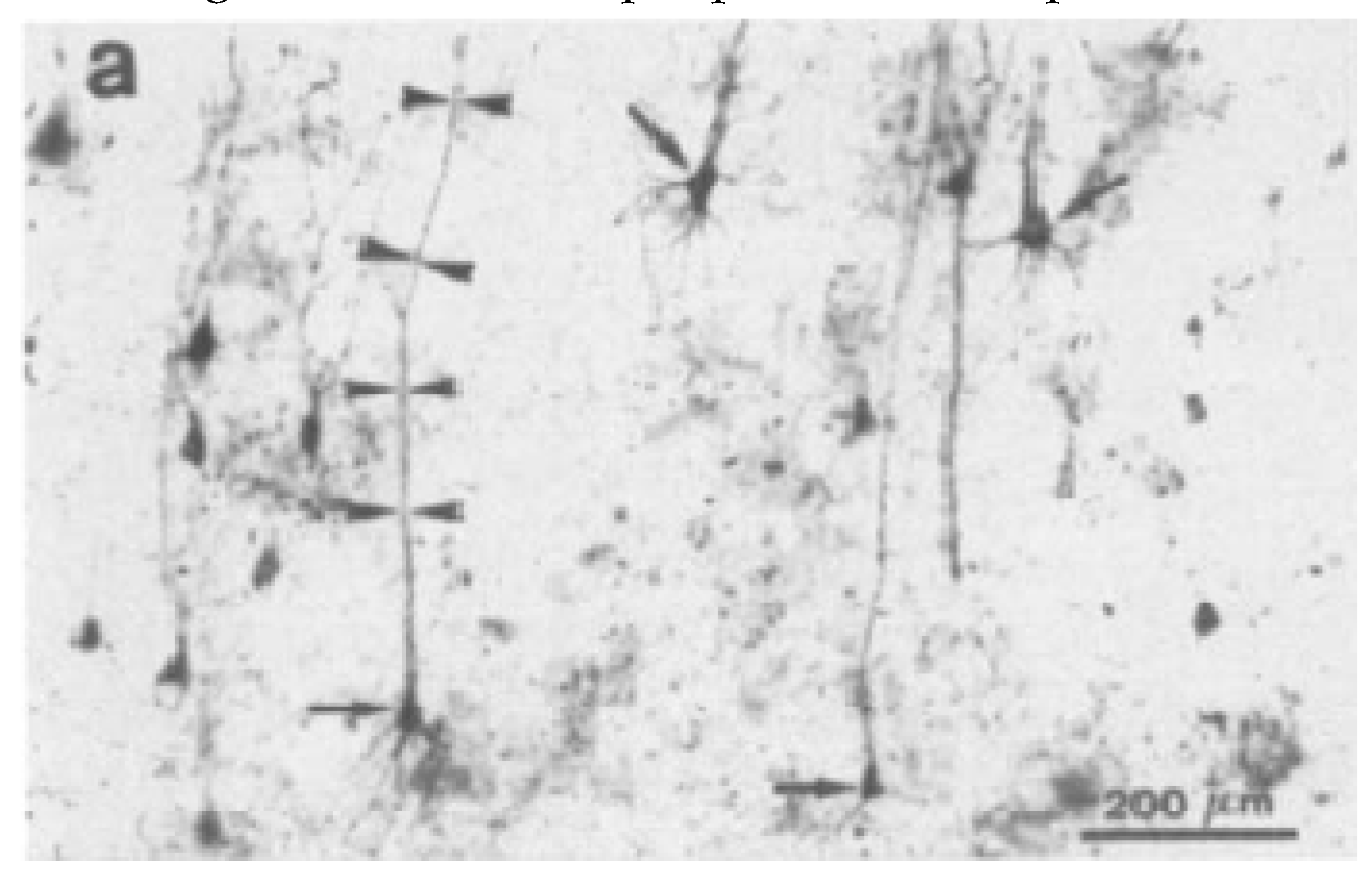
| Canine Cognitive Dysfunction (CCD) | Dog | Cortical atrophy Dysfunction in the neurotransmitter systems Increased oxidative damage Extracellular deposition of diffuse Aβ Neuronal loss Decreased neurogenesis Tau abnormalities, but not NFTs Ventricular enlargement Oligomers of Aβ in the CSF | Loss of cognition and recognition Loss of house training Disorientation Changes in their sleep-wake cycle | [74,75,76,77,78,79,80,81,82,84,85,86] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gołaszewska, A.; Bik, W.; Motyl, T.; Orzechowski, A. Bridging the Gap between Alzheimer’s Disease and Alzheimer’s-like Diseases in Animals. Int. J. Mol. Sci. 2019, 20, 1664. https://doi.org/10.3390/ijms20071664
Gołaszewska A, Bik W, Motyl T, Orzechowski A. Bridging the Gap between Alzheimer’s Disease and Alzheimer’s-like Diseases in Animals. International Journal of Molecular Sciences. 2019; 20(7):1664. https://doi.org/10.3390/ijms20071664
Chicago/Turabian StyleGołaszewska, Anita, Wojciech Bik, Tomasz Motyl, and Arkadiusz Orzechowski. 2019. "Bridging the Gap between Alzheimer’s Disease and Alzheimer’s-like Diseases in Animals" International Journal of Molecular Sciences 20, no. 7: 1664. https://doi.org/10.3390/ijms20071664
APA StyleGołaszewska, A., Bik, W., Motyl, T., & Orzechowski, A. (2019). Bridging the Gap between Alzheimer’s Disease and Alzheimer’s-like Diseases in Animals. International Journal of Molecular Sciences, 20(7), 1664. https://doi.org/10.3390/ijms20071664