Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes?

 ,
,  ,
,  ,
, 

Abstract
:1. Introduction
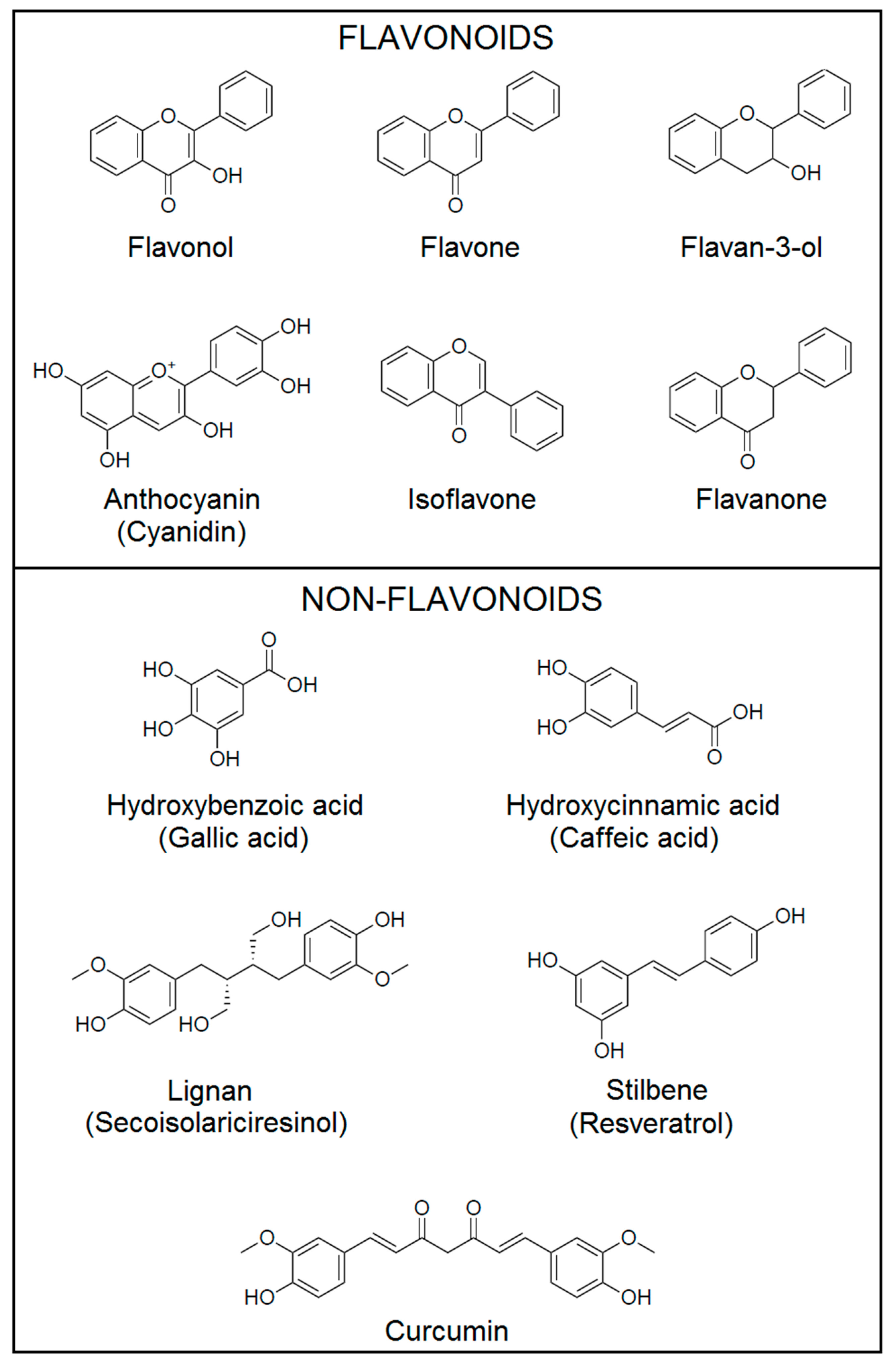
2. Classification of Polyphenols
3. Tumor Immune Microenvironment
3.1. Molecular Mechanisms of Cancer Immunoevasion and Immunosuppression
3.1.1. Reduced Antigenicity and Immunogenicity
3.1.2. Cytokines and Enzymatic Immunosuppression
3.1.3. Suppressive Immune Cells
3.1.4. Exosomes
3.2. Active and Passive Immunotherapy: Basic Concepts and Mechanisms of Action
3.2.1. Therapeutic Cancer Vaccines
Proteins/Peptides Vaccines
Genetic Vaccines
Cell-Based Vaccines
3.2.2. Cytokine Therapy
3.2.3. Co-Stimulatory Receptors Therapy
3.2.4. Adoptive T Cell Transfer Therapy
TILs
TCRs
3.2.5. Immune Checkpoint Inhibitors
CTLA-4
PD-1/PD-L1
Treg cells
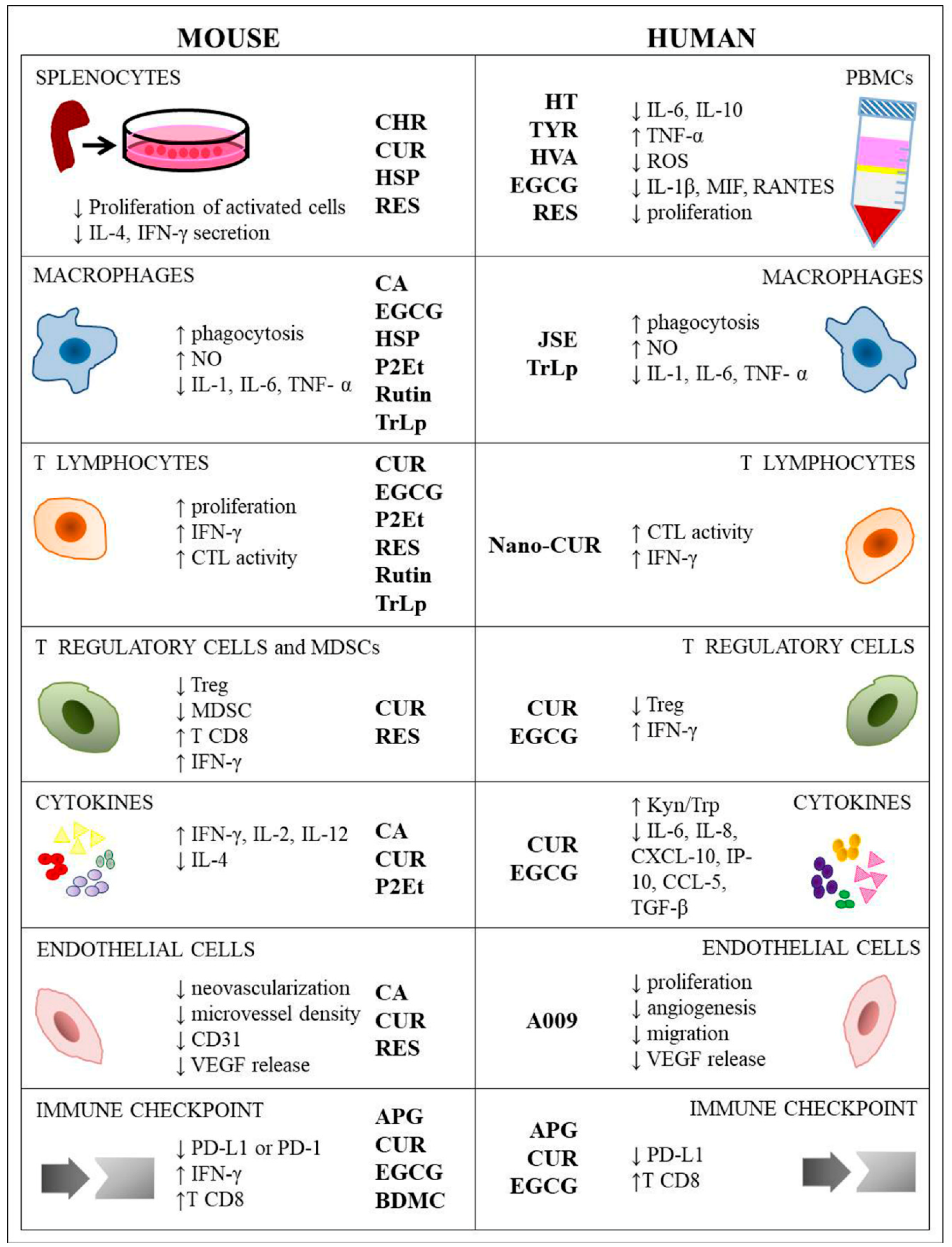
4. Polyphenols and Immune Cells Modulation
4.1. Peripheral Blood Mononuclear Cell (PBMCs) and Murine Splenocytes
4.2. Macrophages
4.3. T Cells
4.4. Treg Cells and MDSCs
4.5. Cytokines
4.6. Endothelial Cells
4.7. Immune Checkpoint
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols. Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Manach, C.; Morand, C.; Rémésy, C.; Jiménez, L. Dietary polyphenols and the prevention of diseases. Crit. Rev. Food Sci. Nutr. 2005, 45, 287–306. [Google Scholar] [CrossRef]
- Marzocchella, L.; Fantini, M.; Benvenuto, M.; Masuelli, L.; Tresoldi, I.; Modesti, A.; Bei, R. Dietary flavonoids: Molecular mechanisms of action as anti-inflammatory agents. Recent Patent Inflamm. Allergy Drug Disc. 2011, 5, 200–220. [Google Scholar] [CrossRef]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef]
- Benvenuto, M.; Fantini, M.; Masuelli, L.; De Smaele, E.; Zazzeroni, F.; Tresoldi, I.; Calabrese, G.; Galvano, F.; Modesti, A.; Bei, R. Inhibition of ErbB receptors, Hedgehog and NF-kappaB signaling by polyphenols in cancer. Front. Biosci. 2013, 18, 1290–1310. [Google Scholar]
- Fantini, M.; Benvenuto, M.; Masuelli, L.; Frajese, G.V.; Tresoldi, I.; Modesti, A.; Bei, R. In vitro and in vivo antitumoral effects of combinations of polyphenols, or polyphenols and anticancer drugs: Perspectives on cancer treatment. Int. J. Mol. Sci. 2015, 16, 9236–9282. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Mattera, R.; Taffera, G.; Giganti, M.G.; Lido, P.; Masuelli, L.; Modesti, A.; Bei, R. The potential protective effects of polyphenols in asbestos-mediated inflammation and carcinogenesis of mesothelium. Nutrients 2016, 8, E275. [Google Scholar] [CrossRef]
- Bei, R.; Masuelli, L.; Turriziani, M.; Li Volti, G.; Malaguarnera, M.; Galvano, F. Impaired expression and function of signaling pathway enzymes by anthocyanins: Role on cancer prevention and progression. Curr. Enzym. Inhib. 2009, 5, 184–197. [Google Scholar] [CrossRef]
- Mattera, R.; Benvenuto, M.; Giganti, M.G.; Tresoldi, I.; Pluchinotta, F.R.; Bergante, S.; Tettamanti, G.; Masuelli, L.; Manzari, V.; Modesti, A.; et al. Effects of polyphenols on oxidative stress-mediated injury in cardiomyocytes. Nutrients 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Gupta, S.C.; Nabavizadeh, A.; Aggarwal, B.B. Regulation of cell signaling pathways by dietary agents for cancer prevention and treatment. Semin. Cancer Biol. 2017, 46, 158–181. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Masuelli, L.; Tresoldi, I.; Sacchetti, P.; Modesti, A.; Galvano, F.; Bei, R. The effects of dietary flavonoids on the regulation of redox inflammatory networks. Front. Biosci. 2012, 17, 2396–2418. [Google Scholar] [CrossRef]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef]
- Visioli, F.; de La Lastra, C.A.; Andres-Lacueva, C.; Aviram, M.; Calhau, C.; Cassano, A.; D’Archivio, M.; Faria, A.; Favé, G.; Fogliano, V.; et al. Polyphenols and human health: A prospectus. Crit. Rev. Food Sci. Nutr. 2011, 51, 524–546. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G. The immune response to tumors. Curr. Protoc. Immunol. 2009. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Monjazeb, A.M.; Zamora, A.E.; Grossenbacher, S.K.; Mirsoian, A.; Sckisel, G.D.; Murphy, W.J. Immunoediting and antigen loss: Overcoming the achilles heel of immunotherapy with antigen non-specific therapies. Front. Oncol. 2013, 3, 197. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35 (Suppl.), S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.G.; Somer, R.A. Intratumor heterogeneity: Novel approaches for resolving genomic architecture and clonal evolution. Mol. Cancer Res. 2017, 15, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Heusschen, R.; Griffioen, A.W.; Thijssen, V.L. Galectin-9 in tumor biology: A jack of multiple trades. Biochim. Biophys. Acta 2013, 1836, 177–185. [Google Scholar] [CrossRef]
- Werb, Z.; Lu, P. The role of stroma in tumor development. Cancer J. 2015, 21, 250–253. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef]
- Komai, T.; Inoue, M.; Okamura, T.; Morita, K.; Iwasaki, Y.; Sumitomo, S.; Shoda, H.; Yamamoto, K.; Fujio, K. Transforming growth factor-beta and Interleukin-10 synergistically regulate humoral immunity via modulating metabolic signals. Front. Immunol. 2018, 9, 1364. [Google Scholar] [CrossRef]
- Yoshimura, A.; Muto, G. TGF-beta function in immune suppression. Curr. Top. Microbiol. Immunol. 2011, 350, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory activity of VEGF in cancer. Int. Rev. Cell. Mol. Biol. 2017, 330, 295–342. [Google Scholar] [CrossRef]
- Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.S.; Firek, A.F.; Langridge, W.H. The role of indoleamine 2,3-Dioxygenase in immune suppression and autoimmunity. Vaccine (Basel) 2015, 3, 703–729. [Google Scholar] [CrossRef]
- Timosenko, E.; Hadjinicolaou, A.V.; Cerundolo, V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy 2017, 9, 83–97. [Google Scholar] [CrossRef]
- Crespo, F.A.; Sun, X.; Cripps, J.G.; Fernandez-Botran, R. The immunoregulatory effects of gangliosides involve immune deviation favoring type-2 T cell responses. J. Leukoc. Biol. 2006, 79, 586–595. [Google Scholar] [CrossRef]
- Krengel, U.; Bousquet, P.A. Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front. Immunol. 2014, 5, 325. [Google Scholar] [CrossRef]
- Liu, M.; Guo, F. Recent updates on cancer immunotherapy. Precis. Clin. Med. 2018, 1, 65–74. [Google Scholar] [CrossRef]
- Frydrychowicz, M.; Boruczkowski, M.; Kolecka-Bednarczyk, A.; Dworacki, G. The dual role of Treg in cancer. Scand. J. Immunol. 2017, 86, 436–443. [Google Scholar] [CrossRef]
- Pere, H.; Tanchot, C.; Bayry, J.; Terme, M.; Taieb, J.; Badoual, C.; Adotevi, O.; Merillon, N.; Marcheteau, E.; Quillien, V.R.; et al. Comprehensive analysis of current approaches to inhibit regulatory T cells in cancer. Oncoimmunology 2012, 1, 326–333. [Google Scholar] [CrossRef]
- Wang, M.; Yin, B.; Wang, H.Y.; Wang, R.F. Current advances in T-cell-based cancer immunotherapy. Immunotherapy 2014, 6, 1265–1278. [Google Scholar] [CrossRef]
- Ward-Hartstonge, K.A.; Kemp, R.A. Regulatory T-cell heterogeneity and the cancer immune response. Clin. Transl. Immunol. 2017, 6, e154. [Google Scholar] [CrossRef]
- Li, X.; Zheng, Y. Regulatory T cell identity: Formation and maintenance. Trends Immunol. 2015, 36, 344–353. [Google Scholar] [CrossRef]
- Goldstein, J.D.; Burlion, A.; Zaragoza, B.; Sendeyo, K.; Polansky, J.K.; Huehn, J.; Piaggio, E.; Salomon, B.L.; Marodon, G. Inhibition of the JAK/STAT signaling pathway in regulatory T cells reveals a very dynamic regulation of Foxp3 expression. PLoS ONE 2016, 11, e0153682. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel Is crucial for the regulatory T cell immune checkpoint in cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of Myeloid-Derived suppressor cells (MDSC) in cancer progression. Vaccines (Basel) 2016, 4, 36. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of Myeloid-Derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Chang, K.; Song, J.Y.; Lim, D.S. Tolerogenic dendritic cell-based immunotherapy. Oncotarget 2017, 8, 90630–90631. [Google Scholar] [CrossRef]
- Ma, Y.; Shurin, G.V.; Gutkin, D.W.; Shurin, M.R. Tumor associated regulatory dendritic cells. Semin. Cancer Biol. 2012, 22, 298–306. [Google Scholar] [CrossRef]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef]
- Pan, Y.; Lei, Z.; Wei, X.; Hao, W. TAK1 deficiency in dendritic cells inhibits adaptive immunity in SRBC-immunized C57BL/6 mice. FEBS Open Bio 2016, 6, 548–557. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Cao, X. The exosomes in tumor immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef]
- Hirayama, M.; Nishimura, Y. The present status and future prospects of peptide-based cancer vaccines. Int. Immunol. 2016, 28, 319–328. [Google Scholar] [CrossRef]
- Mohammed, S.; Bakshi, N.; Chaudri, N.; Akhter, J.; Akhtar, M. Cancer vaccines: Past, present, and future. Adv. Anat. Pathol. 2016, 23, 180–191. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, C.D.; Wu, X.H. Therapeutic cancer vaccines: From initial findings to prospects. Immunol. Lett. 2018, 196, 11–21. [Google Scholar] [CrossRef]
- Yang, X.; Huang, B.; Deng, L.; Hu, Z. Progress in gene therapy using oncolytic vaccinia virus as vectors. J. Cancer Res. Clin. Oncol. 2018, 144, 2433–2440. [Google Scholar] [CrossRef]
- Tian, H.; Shi, G.; Wang, Q.; Li, Y.; Yang, Q.; Li, C.; Yang, G.; Wu, M.; Xie, Q.; Zhang, S.; et al. A novel cancer vaccine with the ability to simultaneously produce anti-PD-1 antibody and GM-CSF in cancer cells and enhance Th1-biased antitumor immunity. Signal Transduct. Target Ther. 2016, 1, 16025. [Google Scholar] [CrossRef]
- Tykocinski, M.L.; Chen, A.; Huang, J.H.; Weber, M.C.; Zheng, G. New designs for cancer vaccine and artificial veto cells: An emerging palette of protein paints. Immunol. Res. 2003, 27, 565–574. [Google Scholar] [CrossRef]
- Sheng, J.; Qin, H.; Zhang, D.; Zhang, X.; Liu, L.; Li, B. New strategies for therapeutic cancer vaccines. Anticancer Agents Med. Chem. 2018. [Google Scholar] [CrossRef]
- Renrick, A.N.; Dunbar, Z.T.; Shanker, A. Update on the current revolution in cancer immunotherapy. Immunotherapy 2019, 1, 15–20. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011, 3, 3856–3893. [Google Scholar] [CrossRef]
- Lo Presti, E.; Pizzolato, G.; Gulotta, E.; Cocorullo, G.; Gulotta, G.; Dieli, F.; Meraviglia, S. Current Advances in gammadelta T Cell-Based tumor immunotherapy. Front. Immunol. 2017, 8, 1401. [Google Scholar] [CrossRef]
- Sasada, T.; Suekane, S. Variation of tumor-infiltrating lymphocytes in human cancers: Controversy on clinical significance. Immunotherapy 2011, 3, 1235–1251. [Google Scholar] [CrossRef]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Model. Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef]
- Hurwitz, A.A.; Cuss, S.M.; Stagliano, K.E.; Zhu, Z. T cell avidity and tumor immunity: Problems and solutions. Cancer Microenviron. 2014, 7, 1–9. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell therapy: A new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Valk, E.; Rudd, C.E.; Schneider, H. CTLA-4 trafficking and surface expression. Trends Immunol. 2008, 29, 272–279. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2017, 7, e1364828. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Serra, G.; Deiana, M.; Spencer, J.P.E.; Corona, G. Olive oil phenolics prevent oxysterol-induced proinflammatory cytokine secretion and Reactive Oxygen Species production in human peripheral blood mononuclear cells, through modulation of p38 and JNK pathways. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Soto, B.L.; Hank, J.A.; Darjatmoko, S.R.; Polans, A.S.; Yanke, E.M.; Rakhmilevich, A.L.; Seo, S.; Kim, K.; Reisfeld, R.A.; Gillies, S.D.; et al. Anti-tumor and immunomodulatory activity of resveratrol in vitro and its potential for combining with cancer immunotherapy. Int. Immunopharmacol. 2011, 11, 1877–1886. [Google Scholar] [CrossRef]
- Chang, M.Y.; Shen, Y.L. Linalool exhibits cytotoxic effects by activating antitumor immunity. Molecules 2014, 19, 6694–6706. [Google Scholar] [CrossRef]
- Sassi, A.; Mokdad Bzéouich, I.; Mustapha, N.; Maatouk, M.; Ghedira, K.; Chekir-Ghedira, L. Immunomodulatory potential of hesperetin and chrysin through the cellular and humoral response. Eur. J. Pharmacol. 2017, 812, 91–96. [Google Scholar] [CrossRef]
- Gao, X.; Deeb, D.; Media, J.; Divine, G.; Jiang, H.; Chapman, R.A.; Gautam, S.C. Immunomodulatory activity of resveratrol: Discrepant in vitro and in vivo immunological effects. Biochem. Pharmacol. 2003, 66, 2427–2435. [Google Scholar] [CrossRef]
- Saleh, F.; Raghupathy, R.; Asfar, S.; Oteifa, M.; Al-Saleh, N. Analysis of the effect of the active compound of green tea (EGCG) on the proliferation of peripheral blood mononuclear cells. BMC Complement. Altern. Med. 2014, 14, 322. [Google Scholar] [CrossRef]
- Huang, A.C.; Cheng, H.Y.; Lin, T.S.; Chen, W.H.; Lin, J.H.; Lin, J.J.; Lu, C.C.; Chiang, J.H.; Hsu, S.C.; Wu, P.P.; et al. Epigallocatechin gallate (EGCG), influences a murine WEHI-3 leukemia model in vivo through enhancing phagocytosis of macrophages and populations of T- and B-cells. In Vivo 2013, 27, 627–634. [Google Scholar]
- Jang, J.Y.; Lee, J.K.; Jeon, Y.K.; Kim, C.W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef]
- Lin, J.P.; Yang, J.S.; Lu, C.C.; Chiang, J.H.; Wu, C.L.; Lin, J.J.; Lin, H.L.; Yang, M.D.; Liu, K.C.; Chiu, T.H.; et al. Rutin inhibits the proliferation of murine leukemia WEHI-3 cells in vivo and promotes immune response in vivo. Leuk. Res. 2009, 33, 823–828. [Google Scholar] [CrossRef]
- Oršolić, N.; Kunštić, M.; Kukolj, M.; Gračan, R.; Nemrava, J. Oxidative stress, polarization of macrophages and tumour angiogenesis: Efficacy of caffeic acid. Chem. Biol. Interact. 2016, 256, 111–124. [Google Scholar] [CrossRef]
- Alonso-Castro, A.J.; Ortiz-Sánchez, E.; Domínguez, F.; Arana-Argáez, V.; Juárez-Vázquez Mdel, C.; Chávez, M.; Carranza-Álvarez, C.; Gaspar-Ramírez, O.; Espinosa-Reyes, G.; López-Toledo, G.; et al. Antitumor and immunomodulatory effects of Justicia spicigera Schltdl (Acanthaceae). J. Ethnopharmacol. 2012, 141, 888–894. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hussaini, R.; White, R.; Atwi, D.; Fried, A.; Sampat, S.; Piao, L.; Pan, Q.; Banerjee, P. TriCurin, a synergistic formulation of curcumin, resveratrol, and epicatechin gallate, repolarizes tumor-associated macrophages and triggers an immune response to cause suppression of HPV+ tumors. Cancer Immunol. Immunother. 2018, 67, 761–774. [Google Scholar] [CrossRef]
- Mukherjee, S.; Baidoo, J.N.E.; Sampat, S.; Mancuso, A.; David, L.; Cohen, L.S.; Zhou, S.; Banerjee, P. Liposomal TriCurin, a synergistic combination of curcumin, epicatechin gallate and resveratrol, repolarizes tumor-associated microglia/macrophages, and eliminates glioblastoma (GBM) and GBM stem cells. Molecules 2018, 23, 201. [Google Scholar] [CrossRef]
- Sharma, S.; Chopra, K.; Kulkarni, S.K.; Agrewala, J.N. Resveratrol and curcumin suppress immune response through CD28/CTLA-4 and CD80 co-stimulatory pathway. Clin. Exp. Immunol. 2007, 147, 155–163. [Google Scholar] [CrossRef]
- Noh, K.T.; Chae, S.H.; Chun, S.H.; Jung, I.D.; Kang, H.K.; Park, Y.M. Resveratrol suppresses tumor progression via the regulation of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2013, 431, 348–353. [Google Scholar] [CrossRef]
- Gualdoni, G.A.; Fuchs, D.; Zlabinger, G.J.; Gostner, J.M. Resveratrol intake enhances indoleamine-2,3-dioxygenase activity in humans. Pharmacol. Rep. 2016, 68, 1065–1068. [Google Scholar] [CrossRef]
- Lasso, P.; Gomez-Cadena, A.; Urueña, C.; Donda, A.; Martinez-Usatorre, A.; Barreto, A.; Romero, P.; Fiorentino, S. Prophylactic vs. therapeutic treatment with P2Et polyphenol-rich extract has opposite effects on tumor growth. Front. Oncol. 2018, 8, 356. [Google Scholar] [CrossRef]
- Gomez-Cadena, A.; Urueña, C.; Prieto, K.; Martinez-Usatorre, A.; Donda, A.; Barreto, A.; Romero, P.; Fiorentino, S. Immune-system-dependent anti-tumor activity of a plant-derived polyphenol rich fraction in a melanoma mouse model. Cell Death Dis. 2016, 7, e2243. [Google Scholar] [CrossRef]
- Chen, L.; Yang, S.; Liao, W.; Xiong, Y. Modification of antitumor immunity and tumor microenvironment by resveratrol in mouse renal tumor model. Cell. Biochem. Biophys. 2015, 72, 617–625. [Google Scholar] [CrossRef]
- Milano, F.; Mari, L.; van de Luijtgaarden, W.; Parikh, K.; Calpe, S.; Krishnadath, K.K. Nanocurcumin inhibits proliferation of esophageal adenocarcinoma cells and enhances the T cell mediated immune response. Front. Oncol. 2013, 3, 137. [Google Scholar] [CrossRef]
- Lu, Y.; Miao, L.; Wang, Y.; Xu, Z.; Zhao, Y.; Shen, Y.; Xiang, G.; Huang, L. Curcumin micelles remodel tumor microenvironment and enhance vaccine activity in an advanced melanoma model. Mol. Ther. 2016, 24, 364–374. [Google Scholar] [CrossRef]
- Focaccetti, C.; Bruno, A.; Magnani, E.; Bartolini, D.; Principi, E.; Dallaglio, K.; Bucci, E.O.; Finzi, G.; Sessa, F.; Noonan, D.M.; et al. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS ONE 2015, 10, e0115686. [Google Scholar] [CrossRef]
- Masuelli, L.; Granato, M.; Benvenuto, M.; Mattera, R.; Bernardini, R.; Mattei, M.; D’Amati, G.; D’Orazi, G.; Faggioni, A.; Bei, R.; et al. Chloroquine supplementation increases the cytotoxic effect of curcumin against Her2/neu overexpressing breast cancer cells. Oncoimmunology 2017, 6, e1356151. [Google Scholar] [CrossRef]
- Masuelli, L.; Benvenuto, M.; Di Stefano, E.; Mattera, R.; Fantini, M.; De Feudis, G.; De Smaele, E.; Tresoldi, I.; Giganti, M.G.; Modesti, A.; et al. Curcumin blocks autophagy and activates apoptosis of malignant mesothelioma cell lines and increases the survival of mice intraperitoneally transplanted with a malignant mesothelioma cell line. Oncotarget 2017, 8, 34405–34422. [Google Scholar] [CrossRef]
- Masuelli, L.; Di Stefano, E.; Fantini, M.; Mattera, R.; Benvenuto, M.; Marzocchella, L.; Sacchetti, P.; Focaccetti, C.; Bernardini, R.; Tresoldi, I.; et al. Resveratrol potentiates the in vitro and in vivo anti-tumoral effects of curcumin in head and neck carcinomas. Oncotarget 2014, 5, 10745–10762. [Google Scholar] [CrossRef]
- Liao, F.; Liu, L.; Luo, E.; Hu, J. Curcumin enhances anti-tumor immune response in tongue squamous cell carcinoma. Arch. Oral Biol. 2018, 92, 32–37. [Google Scholar] [CrossRef]
- Luo, F.; Song, X.; Zhang, Y.; Chu, Y. Low-dose curcumin leads to the inhibition of tumor growth via enhancing CTL-mediated antitumor immunity. Int. Immunopharmacol. 2011, 11, 1234–1240. [Google Scholar] [CrossRef]
- Kim, G.Y.; Kim, K.H.; Lee, S.H.; Yoon, M.S.; Lee, H.J.; Moon, D.O.; Lee, C.M.; Ahn, S.C.; Park, Y.C.; Park, Y.M. Curcumin inhibits immunostimulatory function of dendritic cells: MAPKs and translocation of NF-kappa B as potential targets. J. Immunol. 2005, 174, 8116–8124. [Google Scholar] [CrossRef]
- Yang, Y.; Paik, J.H.; Cho, D.; Cho, J.A.; Kim, C.W. Resveratrol induces the suppression of tumor-derived CD4+CD25+ regulatory T cells. Int. Immunopharmacol. 2008, 8, 5427. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Trung, L.Q.; Inaoka, P.T.; Yamada, K.; An, D.T.; Mizuno, S.; Nakao, S.; Takami, A. The repeated administration of resveratrol has measurable effects on circulating T-cell subsets in humans. Oxid. Med. Cell. Longev. 2017, 2017, 6781872. [Google Scholar] [CrossRef]
- Liu, D.; You, M.; Xu, Y.; Li, F.; Zhang, D.; Li, X.; Hou, Y. Inhibition of curcumin on myeloid-derived suppressor cells is requisite for controlling lung cancer. Int. Immunopharmacol. 2016, 39, 265–272. [Google Scholar] [CrossRef]
- Zou, J.Y.; Su, C.H.; Luo, H.H.; Lei, Y.Y.; Zeng, B.; Zhu, H.S.; Chen, Z.G. Curcumin converts Foxp3+ regulatory T cells to T helper 1 cells in patients with lung cancer. J. Cell. Biochem. 2018, 119, 1420–1428. [Google Scholar] [CrossRef]
- Xu, B.; Yu, L.; Zhao, L.Z. Curcumin up regulates T helper 1 cells in patients with colon cancer. Am. J. Transl. Res. 2017, 9, 1866–1875. [Google Scholar]
- D’Arena, G.; Simeon, V.; De Martino, L.; Statuto, T.; D’Auria, F.; Volpe, S.; Deaglio, S.; Maidecchi, A.; Mattoli, L.; Mercati, V.; et al. Regulatory Tcell modulation by green tea in chronic lymphocytic leukemia. Int. J. Immunopathol. Pharmacol. 2013, 26, 117–125. [Google Scholar] [CrossRef]
- Bergman, M.; Levin, G.S.; Bessler, H.; Djaldetti, M.; Salman, H. Resveratrol affects the cross talk between immune and colon cancer cells. Biomed. Pharmacother. 2013, 67, 43–47. [Google Scholar] [CrossRef]
- Falah, R.R.; Talib, W.H.; Shbailat, S.J. Combination of metformin and curcumin targets breast cancer in mice by angiogenesis inhibition, immune system modulation and induction of p53 independent apoptosis. Ther. Adv. Med. Oncol. 2017, 9, 235–252. [Google Scholar] [CrossRef]
- Mukherjee, S.; Siddiqui, M.A.; Dayal, S.; Ayoub, Y.Z.; Malathi, K. Epigallocatechin-3-gallate suppresses proinflammatory cytokines and chemokines induced by Toll-like receptor 9 agonists in prostate cancer cells. J. Inflamm. Res. 2014, 7, 89–101. [Google Scholar] [CrossRef]
- Yusuf, N.; Nasti, T.H.; Meleth, S.; Elmets, C.A. Resveratrol enhances cell-mediated immune response to DMBA through TLR4 and prevents DMBA induced cutaneous carcinogenesis. Mol. Carcinog. 2009, 48, 713–723. [Google Scholar] [CrossRef]
- Guan, H.; Singh, N.P.; Singh, U.P.; Nagarkatti, P.S.; Nagarkatti, M. Resveratrol prevents endothelial cells injury in high-dose interleukin-2 therapy against melanoma. PLoS ONE 2012, 7, e35650. [Google Scholar] [CrossRef]
- Rossi, T.; Bassani, B.; Gallo, C.; Maramotti, S.; Noonan, D.M.; Albini, A.; Bruno, A. Effect of a purified extract of olive mill waste water on endothelial cell proliferation, apoptosis, migration and capillary-like structure in vitro and in vivo. J. Bioanal. Biomed. 2015, 12, 6. [Google Scholar] [CrossRef]
- Bassani, B.; Rossi, T.; De Stefano, D.; Pizzichini, D.; Corradino, P.; Macrì, N.; Noonan, D.M.; Albini, A.; Bruno, A. Potential chemopreventive activities of a polyphenol rich purifified extract from olive mill wastewater on colon cancer cells. J. Funct. Foods 2016, 27, 236–248. [Google Scholar] [CrossRef]
- Baci, D.; Gallazzi, M.; Cascini, C.; Tramacere, M.; De Stefano, D.; Bruno, A.; Noonan, D.M.; Albini, A. Downregulation of pro-inflammatory and pro-angiogenic pathways in prostate cancer cells by a polyphenol-rich extract from olive mill wastewater. Int. J. Mol. Sci. 2019, 20, 307. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Tian, K.; Chen, X.; Zhang, R.; Mu, X.; Wu, Y.; Wang, D.; Wang, S.; Liu, F.; et al. Apigenin suppresses PD-L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J. Exp. Clin. Cancer Res. 2018, 37, 261. [Google Scholar] [CrossRef]
- Lucas, J.; Hsieh, T.C.; Halicka, H.D.; Darzynkiewicz, Z.; Wu, J.M. Upregulation of PD-L1 expression by resveratrol and piceatannol in breast and colorectal cancer cells occursvia HDAC3/p300-mediated NF-κB signaling. Int. J. Oncol. 2018, 53, 1469–1480. [Google Scholar] [CrossRef]
- Rawangkan, A.; Wongsirisin, P.; Namiki, K.; Iida, K.; Kobayashi, Y.; Shimizu, Y.; Fujiki, H.; Suganuma, M. Green tea catechin is an alternative immune checkpoint inhibitor that inhibits PD-L1 expression and lung tumor growth. Molecules 2018, 23, 2071. [Google Scholar] [CrossRef]
- Shao, Y.; Zhu, W.; Da, J.; Xu, M.; Wang, Y.; Zhou, J.; Wang, Z. Bisdemethoxycurcumin in combination with α-PD-L1 antibody boosts immune response against bladder cancer. OncoTargets Ther. 2017, 10, 2675–2683. [Google Scholar] [CrossRef]
- Han, X.; Shen, T.; Lou, H. Dietary polyphenols and their biological significance. Int. J. Mol. Sci. 2007, 8, 950–988. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Type | Treatment | In Vitro Model | In Vivo Model | Effect on Immune System | Ref. | |
|---|---|---|---|---|---|---|
| MOUSE | SPLENOCYTES | CHR | ♂ Wistar rat, LPS o lectin-stimulated, 3–25 µM, 48 h | ↓ proliferation (LPS) ↑ proliferation (lectin) | [78] | |
| CUR | ♀ Balb/c, + ConA 1 µg/mL or LPS 5 µg/mL + CUR 1–20 µM, 72 h | ↓ T cell proliferation (ConA) ↓ IL-4, IFN-γ secretion ↓ B cell proliferation (LPS) ↓ IgG1, IgG2 production ↔ viability | [88] | |||
| HSP | ♂ Wistar rat, LPS or lectin-stimulated splenocytes, 3–25 µM, 48 h | ↓ proliferation (LPS) ↓ proliferation (lectin) | [78] | |||
| JSE | ♂ C57BL/6, 1–200 µg/mL, 48 h | ↑ proliferation | [85] | |||
| RES | ♀ Balb/c, + ConA 1 µg/mL or LPS 5 µg/mL + RES 1–20 µM, 72 h | ↓ T cell proliferation (ConA) ↓ IL-4, IFN-γ secretion ↓ B cell proliferation (LPS) ↓ IgG1, IgG2 production ↔ viability | [88] | |||
| IL2 + ConA stimulation | ♀ A/J bearing neuroblastoma (NXS2) s.c., 20 mg p.t./every 3 days | ↔ circulating leukocyte population ↑ tumor infiltrating leukocytes (CD45+) ↓ splenocytes proliferation ↓ ADCC | [76] | |||
| ♂ C3H (H-2k) splenocytes, IL-2 or ConA-stimulated + RES 6, 25–50 µM | ♂ C3H (H-2k) RES p.o. 2 mg/day, 5 days/week, 4 weeks | ↑ proliferation (RES 6.25–12.5 µM) ↓ proliferation (RES 25–50 µM) ↔ body weight ↔ peripheral blood cell count ↔ IFN-γ secretion (ConA-stimulated splenocytes) | [79] | |||
| HUMAN | PERIPHERAL BLOOD MONONUCLEAR CELLS | RES | PBMC healthy donor, 0–60 µM | ↓ IL-6, IL-10; ↑ TNF-α; ↔ IFN-γ, IL-1ra, IL-1b | [109] | |
| HT | PBMC healthy donor, pre-treated HT 0.25–1 µM, 30′ + treated Oxysterols mixture 20 µM, 24 h | ↓ IL-1b, MIF, RANTES ↓ intracellular ROS production ↓ p-JNK1/2 | [75] | |||
| TYR | PBMC healthy donor, pre-treated TYR 0.25–1 µM, 30′ + treated Oxysterols mixture 20 µM, 24 h | ↓ IL-1b, MIF, RANTES ↓ intracellular ROS production ↓p-JNK1/2 | [75] | |||
| HVA | PBMC healthy donor, pre-treated HVA 0.25–1 µM, 30′ + treated Oxysterols mixture 20 µM, 24 h | ↓ IL-1b, MIF, RANTES ↓ intracellular ROS production ↓p-JNK1/2 (1 µM) | [75] | |||
| RES | PBMC healthy donor, PHA stimulated, 1–50 µM | ↓ PBMC proliferation ↓ ADCC | [76] | |||
| PBMC healthy donors, HT29, 0–60 µM | ↓ IL-6, IL-10, TNF-α, IFN-γ, IL-1ra, IL-1b | [109] | ||||
| PBMC healthy donors, RKO, 0–60 µM | ↓ IL-1b, IFN-γ, IL-10 ↔ IL-6 and IL-1ra ↑ TNF-α | [109] | ||||
| Linalool | Lymphocytes healthy donor, 227 µM, 24 h | ↑ CD40-ligand, CD40, IFN-γ, IL-12 p40, IL-13, IL-17F, IL-1β, IL-2, IL-21, IL-21R, IL-23p19, IL-4, IL-6Sr, TNF-α | [77] | |||
| EGCG | Breast cancer patients, PBMC stimulated with PHA, anti-CD3, or Her2/neu and p53 antigen peptides, EGCG 0.125–50 µg/mL | ↓ PBMC proliferation > 10 µg/mL ↓ IFN-γ production > 10 µg/mL | [80] | |||
| MOUSE | MACROPHAGES | CA | ♂ Swiss albino bearing Ehrlich ascites tumor (EAT) cells | ↑ macrophages count ↑ macrophages cytotoxicity ↓ ARG1; ↔ NO ↔ neutrophils, lymphocytes count | [84] | |
| CHR | ♂ Wistar rat, LPS o lectin-stimulated splenocytes, 3–25 µM, 48 h | ↓ macrophage lysosomal enzyme activity ↓ NO production | [78] | |||
| CUR | ♀ Balb/c, peritoneal macrophages, LPS stimulated + 1–20 µM, 48 h | ↓ IL-1, IL-6, TNF-α ↑ IL-10 ↓ CD80, CD86 expression ↔ CD40 expression | [88] | |||
| EGCG | ♂ Balb/c bearing leukemia cells (WEHI-3), 5–40 mg/kg p.o. | ↑ phagocytosis | [81] | |||
| Balb/c bearing mammary cancer cells (4T1) s.c., EGCG i.p. 10 mg/kg, day 7-day 9-day 11 | ↓ infiltration TAM and M2 ↓ CSF-1, CCL-2 ↓ IL-6, TGF-β ↑ TNF-α | [82] | ||||
| HSP | ♂ Wistar rat, LPS o lectin-stimulated splenocytes, HSP 3–25 µM, 48 h | ↑ macrophage lysosomal enzyme activity ↔ NO production | [78] | |||
| P2Et | Melanoma cells (B16F10), 72.7 µg/mL, 36 h | C57BL/6 bearing melanoma cells (B16F10) s.c., 75 mg/mL | ↑ CD45+CD220−CD11c+ ↑ CD86, CD40, MHCII, CD70 ↑ BMDCs phagocytosis | [92] | ||
| ♀ C57BL/6 Healthy, 75 mg/kg i.p., twice/week/3weeks | ↑ DCs, ↑ MDSC-LC | [91] | ||||
| ♀ Balb/c Healthy, 75 mg/kg i.p., twice/week/5 weeks | ↑ DCs ↑ MDSC-LC | [91] | ||||
| RES | ♂ C57BL/6, EG7 cells, BMDC pre-treated 1 h, 20–50 µM + 18 h IFN-γ 100 U/mL | ↓ IDO expression ↓ IRF-1 expression ↓ STAT1, PKCδ | [89] | |||
| ♀ Balb/c, peritoneal macrophages, LPS stimulated + 1–20 µM, 48 h | ↓ IL-1, IL-6, TNF-α ↑ IL-10 ↓ CD80, CD86 expression ↔ CD40 expression | [88] | ||||
| Rutin | ♂ Balb/c bearing leukemia cells (WEHI-3), macrophages from PBMC or peritoneum, 6–12 mg/kg, p.o., 3 weeks | ↑ phagocytosis | [83] | |||
| TrLp | ♂ C57BL/6 bearing Glioblastoma cells (GL261) i.c. implanted, TrLp 1,28 mM, i.p., every 24 h, 5 days | Switch from M2-like TAM ARG1highiNOSlow to M1-like TAM phenotype ARG1lowiNOShigh ↑ NO production | [87] | |||
| C57BL/6 bearing Lung cancer cell (HPV+ TC-1), 64 µM, i.t. infusion every 24 h, 5 days | ‘tumor-core’: E6+ tumor cells, ‘tumor-periphery’: Iba1+ TAM Switch from ARG1highiNOSlowIL-12lowIL-10high M2 TAM to ARG1lowiNOShighIL-12highIL-10low M1 TAM ↑ NO ↓ p-STAT3 ↑ p-STAT1, p-NF-Kb | [86] | ||||
| HUMAN | MACROPHAGES | JSE | JSE, 1–200 µg/mL, 48 h | ↑ phagocytosis, ↑ NO, ↑ H2O2 | [85] | |
| TrLp | HNSCC cells (UMSCC47); Nu/nu mice i.t. TrLp thrice/week/5weeks | Switch from ARG1highiNOSlow Iba1+ M2 Macrophages to Iba1+ TAM ARG1lowiNOShigh M1 ↓ p-STAT3 ↑ p-STAT1, p-NF-kB ↑ NO | [86] | |||
| Nano-CUR | 0–50 µM, 48 h | ↔ CD80, CCR7 ↑ CD86 ↔ TNF-α, IL-6, IL-12 | [94] | |||
| MOUSE | T LYMPHOCYTES | CHR or HSP | ♂ Wistar rat, LPS o lectin-stimulated splenocytes, CHR or HSP 3–25 µM, 48 h | ↑ CTL activity against B16F10 | [78] | |
| CUR | ♀ Balb/c splenocytes, + ConA 1 µg/mL + CUR 1–20 µM | ↓ CD28 expression on CD4 ↑ CTLA-4 expression on CD4 ↓ proliferation T cells ↓ IFN-γ, IL-4 secretion | [88] | |||
| ♀ Balb/c bearing mammary cancer cells (TUBO) s.c., CUR p.o. 2 mg/50 µL oil, 3 days/week, ± CQ 2 mg/50 µL water, 5 days/week | ↑ CD8, ↓ Foxp3 Treg cells ↓ CD8, ↑Foxp3 Treg cells, (CUR+CQ) | [97] | ||||
| ♀ C57BL/6 bearing oral carcinoma 4NQO-induced in drinking water for 16 weeks, CUR 4 weeks | ↑ CD8 in tumor microenvironment | [100] | ||||
| ♀ C57BL/6 bearing Lewis lung carcinoma (LLC), splenic lymphocytes isolation, activation and treatment with CUR 1.5 µg/mL, 48 h | ↑ frequency and number of T cells (CUR < 1.5 µg/mL) ↔ frequency and number of B cells, DCs, NK ↓ frequency and number of T cells (CUR > 2 µg/mL) | [101] | ||||
| ♀ C57BL/6 bearing Lewis lung carcinoma cells (LLC), CUR 0–100 mg/kg/day/mouse, 10 days, i.p. | ↑ CD8 cytotoxicity and proliferation (CUR < 1.5 µg/mL) ↑IFN-γ | [101] | ||||
| ♂ C57BL/6 BMDC LPS-matured, treated CUR 0–25 µM, 45′ | ↓ BMDCs maturation ↓ CD80, CD86, MHCII ↓ IL-1, IL-6, TNF-α ↓ T cell activation ↓ IFN-γ production | [102] | ||||
| EGCG | ♂ Balb/c bearing leukemia (WEHI-3) cells, 5–40 mg/kg p.o. | ↑ CD3+ | [81] | |||
| P2Et | Vaccination with melanoma cells (B16F10) pre-treated with 101.6 µg/mL P2Et, 48 h | ↑ CD8+CD44+, CD8+CD44+CD62L+ ↑ CD8+ IFN-γ+ | [92] | |||
| ♀ C57BL/6 bearing melanoma cells (B16F10), 75 mg/kg, i.p. twice/week/10 days + s.c. twice/week/21 days | ↓ CD3+, CD4+, CD8+ (LN) ↓ CD44+ (LN) | [91] | ||||
| ♀ C57BL/6 Healthy, 75 mg/kg i.p., twice/week/3 weeks | ↑ CD4+, CD8+ | [91] | ||||
| ♀ Balb/c bearing mouse mammary cancer cells (4T1) cells, 75 mg/kg, i.p. twice/week/10 days + s.c. twice/week/32 days | ↓ CD3+, CD4+, CD8+ (LN) ↔ CD44+ (LN) ↓ CD4+ TEM↑ CD8+ TN↓ CD8+ TEM | [91] | ||||
| ♀ Balb/c Healthy,75 mg/kg i.p., twice/week/5 weeks | ↑ CD4+, CD8+ | [91] | ||||
| RES | ♀ Balb/c bearing renal adenocarcinoma cells (RENCA), 1–5 mg/kg, i.p. | ↑ CD8 density; ↑CD69+ ↑ CD8 Perforin, Granzyme B, FasL | [93] | |||
| ♀ C57BL/6 spleen of T cell lymphoma (EG7)-bearing mice, 25–75 µM RES, 24 h | ♀ C57BL/6, i.p. 4 mg/kg RES | ↑ CD8+IFN-γ+ | [103] | |||
| ♂ C57BL/6 OT-1; CD8 co-cultured with DC pulsed with OVA + 18 h IFN-γ 100 U/mL | ↑ CD8 proliferation ↑ CTL activity | [89] | ||||
| ♀ Balb/c splenocytes, ConA, RES 1–20 µM | ↓ CD28 expression on CD4+ ↔ CTLA-4 expression on CD4+ ↓ proliferation T cells ↓ IFN-γ, IL-4 secretion | [88] | ||||
| Rutin | ♂ Balb/c bearing leukemia cells (WEHI-3), macrophages from PBMC or peritoneum, 6–12 mg/kg, p.o., 3 weeks | ↑ CD3, CD19 ↓ CD11b, Mac3 | [83] | |||
| TrLp | C57BL/6 bearing Lung cancer cell (HPV+ TC-1), TrLp 64µM, i.t. infusion/every 24 h/5 days | ↑ CD8+ CTL | [86] | |||
| HUMAN | T LYMPHOCYTES | Nano-CUR | 0–50 µM, 48 h | ↔ phenotype resting T cells ↔ cytokine production in resting T cells ↓ TNF-α, IL-6, IL-8, IL-10, IL-1b in activated T cells | [94] | |
| Oral carcinoma (OE19), 50 µM, 48 h | ↑ CTLs lysis ↔ TNF-α, IL-8, IFN-γ, IL-2 | [94] | ||||
| Oral carcinoma (OE33), 50 µM, 48 h | ↑ CTLs lysis ↑ IFN-γ ↓ IL-8 ↔ TNF-α, IL-2 | [94] | ||||
| MOUSE | T REGULATORY CELLS AND MIELOID DERIVED SUPPRESOR CELLS | CUR | ♀ Balb/c splenocytes, + ConA 1 µg/mL + CUR 1–20 µM | ↔ frequency CD4+ CD25+ Treg cells | [88] | |
| ♀ C57BL/6 bearing oral carcinoma 4NQO-induced in drinking water for 16 weeks, CUR 4 weeks | ↓ Treg cells (CD4+ CD25+ Foxp3+) ↓ MDSCs (CD11b+ GR1+) ↑ CD8+ in tumor microenvironment ↓ PD-L1, p-STAT3 | [100] | ||||
| ♀ C57BL/6 bearing melanoma cells (B16F10) s.c., treated with CUR-PEG and LCP Trp2-based vaccine | ↓ MDSCs ↓ Treg cells ↑ CD8+ | [95] | ||||
| ♀ C57BL/6 bearing Lewis lung carcinoma cells (LLC), 50 mg/kg p.o. daily | ↓ MDSCs ↓ ARG1, iNOS, ROS ↑ F4/80, MHCII, CD80, CD11c ↑ CD8+, CD4+ ↓ IL-6 in tumor microenvironment | [105] | ||||
| P2Et | ♀ C57BL/6 bearing melanoma cells (B16F10) cells, 75 mg/kg, i.p. twice/week/10 days + s.c. twice/week/21 days | ↑ MDSC-LC cells | [91] | |||
| ♀ C57BL/6 Healthy, 75 mg/kg i.p., twice/week/3 weeks | ↑ CTLA-4+, Foxp3+ Treg cells | [91] | ||||
| ♀ Balb/c bearing mammary cancer cells (4T1) cells, 75 mg/kg, i.p. twice/week/10 days + s.c. twice/week/32 days | ↑ MDSC-LC cells | [91] | ||||
| ♀ Balb/c Healthy,75 mg/kg i.p., twice/week/5 weeks | ↑ CTLA-4+, Foxp3+ Treg cells | [91] | ||||
| RES | ♀ Balb/c bearing renal carcinoma cells (RENCA) cells, i.p. 1–5 mg/kg | ↔ frequency | [93] | |||
| ♀ Balb/c bearing renal carcinoma cells (RENCA) cells, i.p. 1–5 mg/kg | ↓ frequency | [93] | ||||
| ♀ Balb/c splenocytes, + ConA 1 µg/mL + RES 1–20 µM | ↔ frequency CD4+ CD25+ Treg cells | [88] | ||||
| ♀ C57BL/6 spleen of T cell lymphoma (EG7)-bearing mice, 25–75 µM RES, 24 h | ♀ C57BL/6 bearing T cell lymphoma (EG7), i.p. 4 mg/kg | ↓ CD4+ CD25+ FoxP3+ ↓ CD4+CD25+/CD4+ ratio ↓ TGF-b ↑ CD8+IFN-γ+ | [103] | |||
| ♀ Balb/c spleen of colon cancer (CT-26)-bearing mice, 25–75 µM, 24 h | ↓ CD4+ CD25+ FoxP3+ ↓ CD4+CD25+/CD4+ ratio | [103] | ||||
| HUMAN | T REGULATORY CELLS | CUR | PBMC from Lung cancer patients treated 3 g/day, 2 weeks | convert (CD4+CD25+Foxp3+) Treg cells into IFN-γ+ Th1 cells | [106] | |
| PBMC from advanced colon carcinoma patients treated 3 g/day, 2 weeks | ↓ Treg cells (CD4+CD25+Foxp3+) ↑ CD4+CD25+Foxp3−, IFN-γ+ | [106] | ||||
| EGCG, GTE | CLL patients (Rai stage 0), 6 months, 4602 mg of green tea leaves, 189 mg EGCG, 97,5 mg caffeine | ↓ circulating lymphocytes and Treg cells ↓ IL-10 and TGF-β | [108] | |||
| RES | Healthy subjects, 1 g/day, 4 weeks | ↑ circulating Treg cells (CD3+CD4+CD25+CD127dim/neg) ↑ γδ+ NKG2D+ T cell ↑ CD3−CD56+ NKG2D+ NK ↔ CD8, CD4, CD19 ↓ TNF-α, MCP-1 | [104] | |||
| MOUSE | CYTOKINES | CA | ♂ Swiss albino bearing Ehrlich ascites tumor (EAT) cells | ↑ IL-2, IL-12, IFN-γ ↓ IL-4 and IL-10 | [84] | |
| CUR | ♀ Balb/c bearing mammary cancer cells (EMT6/P) s.c., Met 100 µL 80 mg/kg i.p. + CUR 100 µL 50 mg/kg p.o. | ↑ IFN-γ, IL-4 ↔ IL-2, IL-10 | [110] | |||
| ♀ C57BL/6 bearing melanoma cells (B16F10) s.c., treated with CUR-PEG and LCP Trp2-based vaccine | ↓ IL-6, CCL2 in tumor microenvironment ↑ TNF-α, IFN-γ in tumor microenvironment ↑ CTL response | [95] | ||||
| P2Et | ♀ C57BL/6 bearing melanoma cells (B16F10) s.c., 75 mg/kg, i.p. twice/week/10 days + s.c. twice/week/21 days | ↑ TNF-α, IL-6 ↓ IL-17, IL-4 | [91] | |||
| ♀ Balb/c bearing mammary cancer cells (4T1) s.c., 75 mg/kg, i.p. twice/week/10 days + s.c. twice/week/32 days | ↑ TNF-α, IL-6 | [91] | ||||
| ♀ C57BL/6 Healthy, 75 mg/kg i.p., twice/week/3 weeks | ↑ IL-10, IL-17, IFN-γ, IL-6, IL-4, IL-2 | [91] | ||||
| ♀ Balb/c Healthy,75 mg/kg i.p., twice/week/5 weeks | ↑ IFN-γ, IL-6 | [91] | ||||
| HUMAN | CYTOKINES | RES | ♂ healthy subject, 5 g, orally administered | ↑ Kynurenin/tryptophan ratio as measure of IDO activity | [90] | |
| EGCG | Prostate cancer cells (PC3, DU145, LnCap), pre-treated EGCG 40 µg/mL, 24 h and then transfected with CpG-ODN 1 µM, 6 or 24 h | ↓ IL-6, IL-8, CXCL-1, IP-10, CCL-5, TGF-β1 | [111] | |||
| MOUSE | ENDOTHELIAL CELLS | CA | ♂ Swiss albino bearing Ehrlich ascites tumor (EAT) cells | ↓ neovascularization ↓ reduced microvessel density ↓ VEGF release in ascite | [84] | |
| CUR | ♀ C57BL/6 bearing melanoma cells (B16F10) s.c., treated with CUR-PEG and LCP Trp2-based vaccine | ↓ tumor vessels ↓ TAF | [95] | |||
| P2Et | Melanoma cells (B16F10), P2Et 1.9–250 µg/mL | ↑ ICD, ↑autophagy | [92] | |||
| RES | ♀ C3/HeN or C3/HeJ (TLR4 mutated) bearing skin cancer DMBA-induced, 10 µmol/mouse, topically applied 1h before DMBA | ↑ IFN-γ+ (skin lysates) ↑ IL-12 (skin lysates) ↓ MMP-2 MMP-9 ↓ VEGF (skin lysates) ↓ CD31 in tumor | [112] | |||
| ♀ C57BL/6 bearing melanoma cells (B16F10) s.c. or i.v., HDIL-2 75.000 U, 3 times/day/3 days + RES 100 mg/kg p.o., 5 days | ↓ vascular leak syndrome | [113] | ||||
| ♀ Balb/c bearing renal carcinoma cells (RENCA) cells, i.p. 1–5 mg/kg | ↑ IFN-γ, Fas ↓ IL-6, IL-10 | [93] | ||||
| HUMAN | ENDOTHELIAL CELLS | A009 | Human umbilical vein endothelial cells (HUVEC) | ↓ proliferation ↓ angiogenesis ↓ migration | [114] | |
| Colon cancer cells (HT-29, HCT-116);Prostate cancer cells (PC-3, DU-145, LNCaP) | ↓ VEGF, IL-8 CXCL8, CXCL12 ↓ proliferation ↓ angiogenesis ↓ migration | [115,116] | ||||
| MOUSE | IMMUNE CHECKPOINT | APG or CUR | ♀ C57BL/6 bearing melanoma cells (B16F10) s.c., CUR 50 mg/kg or APG 150 mg/kg, p.o., 12 days | ↓ PD-L1 ↑ CD4+ CD8+ in tumor microenvironment | [117] | |
| EGCG or GTE | Melanoma cells (B16F10) co-culture with tumor specific T cells, EGCG 30 µM | ♀ A/J mice bearing lung cancer NNK-induced; GTE 0.3% in drinking water | ↓ PD-L1 | [119] | ||
| BDMC | ♀ C57BL/6 bearing bladder cancer cells (MB49) s.c./i.v. treated with Anti-PD-L1 Ab + BDMC 3 mg/kg, 4/2 weeks | ↑ CD8+ (SPL and LN) ↑ IFN-γ, granzyme B ↓ PD-1 ↑ survival | [120] | |||
| HUMAN | IMMUNE CHECKPOINT | APG or CUR | Melanoma cell lines (A375, A2058, RPMI-7951) treated APG 5–60 µM or CUR 5–30 µM, 24 h; Jurkat cell-mediated A375 killing assays | ↓ PD-L1 ↓ p-STAT1 ↑ CD8 citotoxicity ↑ IL-2 | [117] | |
| RES or Pic | Breast cancer cells (Cal51, BT549, BT474) and Colon cancer cells (SKBR3, HCT116, SW480, HT29 and SW620)RES 50 µM, Pic 50 µM | ↑ PD-L1 ↑ DNA damage | [118] | |||
| EGCG or GTE | Lung cancer cells (A549, H1299, Lu99), 50–100 µg/mL | ↓ PD-L1 | [119] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Focaccetti, C.; Izzi, V.; Benvenuto, M.; Fazi, S.; Ciuffa, S.; Giganti, M.G.; Potenza, V.; Manzari, V.; Modesti, A.; Bei, R. Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes? Int. J. Mol. Sci. 2019, 20, 1714. https://doi.org/10.3390/ijms20071714
Focaccetti C, Izzi V, Benvenuto M, Fazi S, Ciuffa S, Giganti MG, Potenza V, Manzari V, Modesti A, Bei R. Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes? International Journal of Molecular Sciences. 2019; 20(7):1714. https://doi.org/10.3390/ijms20071714
Chicago/Turabian StyleFocaccetti, Chiara, Valerio Izzi, Monica Benvenuto, Sara Fazi, Sara Ciuffa, Maria Gabriella Giganti, Vito Potenza, Vittorio Manzari, Andrea Modesti, and Roberto Bei. 2019. "Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes?" International Journal of Molecular Sciences 20, no. 7: 1714. https://doi.org/10.3390/ijms20071714
APA StyleFocaccetti, C., Izzi, V., Benvenuto, M., Fazi, S., Ciuffa, S., Giganti, M. G., Potenza, V., Manzari, V., Modesti, A., & Bei, R. (2019). Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes? International Journal of Molecular Sciences, 20(7), 1714. https://doi.org/10.3390/ijms20071714