Targeting Chemoresistant Tumors: Could TRIM Proteins-p53 Axis Be a Possible Answer?

 ,
, 
Abstract
:1. Introduction
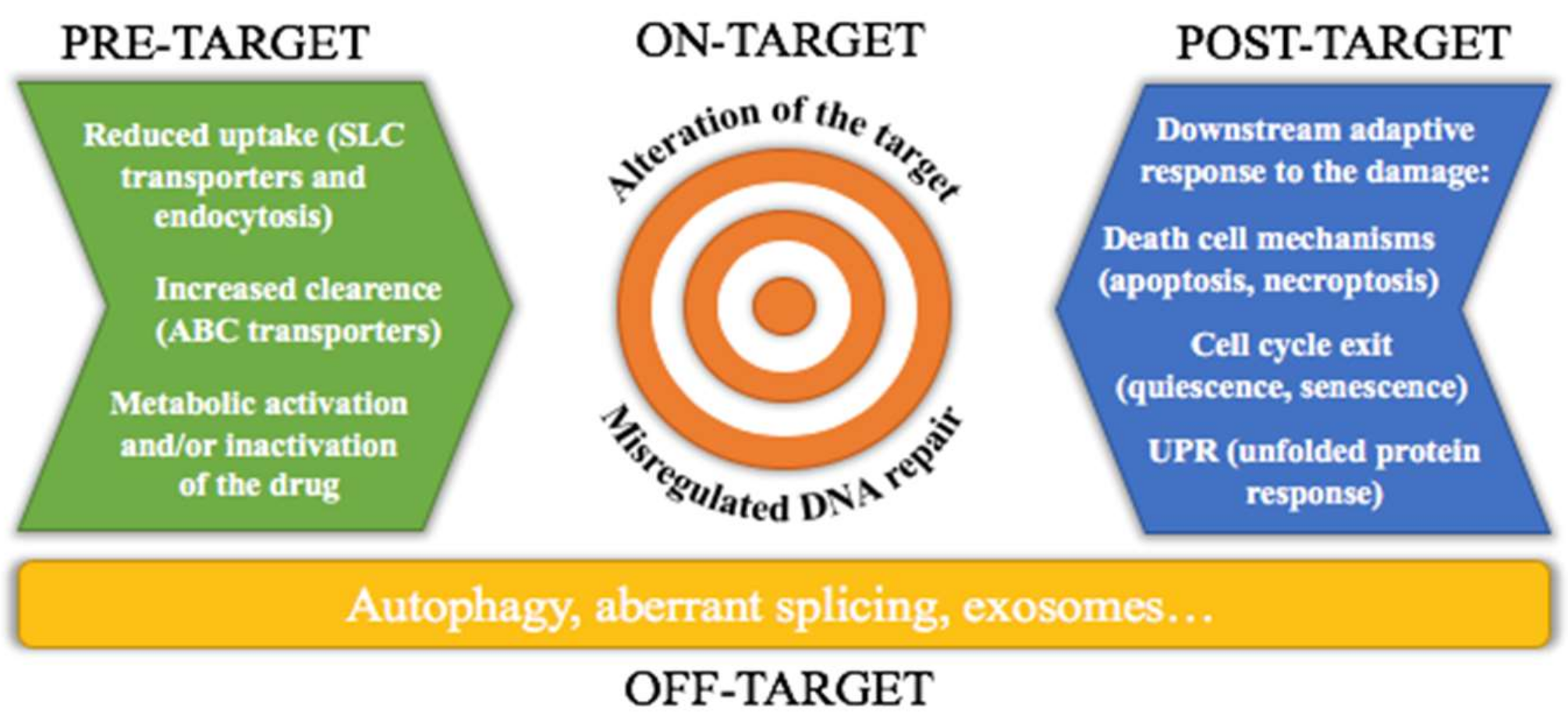
2. Chemoresistance Mechanisms
2.1. Tolerance to DNA Damage and DNA Repair
2.2. Apoptosis and Cell Cycle Regulation
2.3. Autophagy
3. TRIM Proteins as Chemosensitizers
3.1. TRIM Family Proteins Structure and Function
3.2. TRIM Proteins in Cancerogenesis and Chemoresistance
3.2.1. p53 Positive Regulatory TRIM Proteins
3.2.2. p53 Negative Regulatory TRIM Proteins
3.3. p53-Indipendent Resistance Acquisition Mechanisms Correlated to TRIM Proteins
4. TRIM Proteins as Promising Targets for Overcoming Chemotherapy Resistance
5. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TRIM | TRIpartite Motif |
| SLC | SoLute Carrier |
| MDR | MultiDrug Resistance |
| ABC | ATP-Binding Cassette |
| MGMT | O6-MethylGuanine-DNA-MethylTransferase |
| IAP | Inhibitors of Apoptosis Protein |
| CFLAR | CASP8 and FADD like apoptosis regulator |
| EGFR | Epidermal Growth Factor Receptor |
| IGF1R | Insulin-like Growth Factor 1 Receptor |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| NF-κB | Nuclear Factor κB |
| UPR | Unfolded Protein Response |
| PML | ProMyelocytic Leukemia |
| POD | Promyelocytic Oncogenic Domain |
| RARα | Retinoic Acid Receptor α |
| APL | Acute Promyelocytic Leukemia |
| ATC | Anaplastic Thyroid Cancer |
| ccRCC | Clear Cell Renal Cell Carcinoma |
| GMPS | Guanine MonoPhosphate Synthase |
| EMT | Epithelial Mesenchymal Transition |
| TIF1α | Transcription Intermediary Factor 1α |
| APC/C | Anaphase Promoting Complex or Cyclosome |
| IR | Ionizing Radiation |
| ATM | Ataxia Telangiectasia-Mutated |
| Tip60 | Tat-Interactive Protein 60 |
| CIC | Cancer-Initiating Cell |
| NEMO | NF-κB Essential Modulator |
| TRAF | Tumor necrosis factor Receptor Associated Factor |
| TAT | HIV-1 TransActivator of Transcription |
| TBM | TRAF6 Binding Motif |
| PIAS3 | Protein Inhibitor of Activated STAT3 |
| TAK1 | Transforming growth factor β Activated Kinase 1 |
| SOCS1 | Suppressor of Cytokine Signalling 1 |
| PIK3CA | PhosphatidylInositol-4,5-bisphosphate 3-Kinase subunit alpha |
| FASN | Fatty Acid Synthase |
| PAR-4 | Prostate apoptosis response protein 4 |
| EGCG | EpiGalloCatechin Gallate |
References
- Cornelison, R.; Llaneza, D.C.; Landen, C.N. Emerging therapeutics to overcome chemoresistance in epithelial ovarian cancer: A mini-review. Int. J. Mol. Sci. 2017, 18, 2171. [Google Scholar] [CrossRef] [PubMed]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef]
- Salgia, R.; Kulkarni, P. The genetic/non-genetic duality of drug ‘Resistance’ in cancer. Trends Cancer 2018, 4, 110–118. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor evolution as a therapeutic target. Cancer Discov. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Pisco, A.O.; Jackson, D.A.; Huang, S. Reduced intracellular drug accumulation in drug-resistant leukemia cells is not only solely due to MDR-mediated efflux but also to decreased uptake. Front. Oncol. 2014, 4, 306. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Fodale, V.; Pierobon, M.; Liotta, L.; Petricoin, E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer. J. 2011, 17, 89–95. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Anufrieva, K.S.; Shender, V.О.; Arapidi, G.P.; Pavlyukov, M.S.; Shakhparonov, M.I.; Shnaider, P.V.; Butenko, I.O.; Lagarkova, M.A.; Govorun, V.M. Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 2018, 10, 49. [Google Scholar] [CrossRef]
- Sharma, A. Chemoresistance in cancer cells: Exosomes as potential regulators of therapeutic tumor heterogeneity. Nanomedicine 2017, 12, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Fan, C.H.; Liu, W.L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef]
- Hopper-Borge, E.A.; Nasto, R.E.; Ratushny, V.; Weiner, L.M.; Golemis, E.A.; Astsaturov, I. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin. Ther. Targets 2009, 13, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, S.; Mettler, E.; Fottner, C.; Miederer, M.; Kaina, B.; Weber, M.M. The impact of the IGF-1 system of cancer cells on radiation response—An in vitro study. Clin. Transl. Radiat. Oncol. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Salehan, M.R.; Morse, H.R. DNA damage repair and tolerance: A role in chemotherapeutic drug resistance. Br. J. Biomed. Sci. 2013, 70, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Hersey, P.; Zhang, X.D. Overcoming resistance of cancer cells to apoptosis. J. Cell. Physiol. 2003, 196, 9–18. [Google Scholar] [CrossRef]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef]
- Gordon, R.R.; Nelson, P.S. Cellular senescence and cancer chemotherapy resistance. Drug Resist. Updat. 2012, 15, 123–131. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Eberhart, K.; Oral, O.; Gozuacik, D. Chapter 13—Induction of autophagic cell death by anticancer agents. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2014; pp. 179–202. [Google Scholar]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.-M.; Chang, T.-H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Caratozzolo, M.F.; Marzano, F.; Mastropasqua, F.; Sbisà, E.; Tullo, A. TRIM8: Making the right decision between the oncogene and tumour suppressor role. Genes 2017, 8, E354. [Google Scholar] [CrossRef] [PubMed]
- Joazeiro, C.A.; Weissman, A.M. RING finger proteins: Mediators of ubiquitin ligase activity. Cell 2000, 102, 549–552. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Aguilar, R.C.; Wendland, B. Ubiquitin: Not just for proteasomes anymore. Curr. Opin. Cell. Biol. 2003, 15, 184–190. [Google Scholar] [CrossRef]
- Conaway, R.C.; Brower, C.S.; Conaway, J.W. Emerging roles of ubiquitin in transcription regulation. Science 2002, 296, 1254–1258. [Google Scholar] [CrossRef]
- Negorev, D.; Maul, G.G. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 2001, 20, 7234–7242. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J. The role of tripartite motif family proteins in TGF-β signaling pathway and cancer. J. Cancer Prev. 2018, 23, 162–169. [Google Scholar] [CrossRef]
- Li, L.; Qi, Y.; Ma, X.; Xiong, G.; Wang, L.; Bao, C. TRIM22 knockdown suppresses chronic myeloid leukemia via inhibiting PI3K/Akt/mTOR signaling pathway. Cell Biol. Int. 2018, 42, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Khaku, S.; Jennis, M.; Zhou, Y.; Murphy, M.E. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol. Cancer Res. MCR 2015, 13, 250–262. [Google Scholar] [CrossRef]
- Piao, M.Y.; Cao, H.L.; He, N.N.; Xu, M.Q.; Dong, W.X.; Wang, W.Q.; Wang, B.M.; Zhou, B. Potential role of TRIM3 as a novel tumour suppressor in colorectal cancer (CRC) development. Scand. J. Gastroenterol. 2016, 51, 572–582. [Google Scholar] [CrossRef]
- Liu, Y.; Raheja, R.; Yeh, N.; Ciznadija, D.; Pedraza, A.M.; Ozawa, T.; Hukkelhoven, E.; Erdjument-Bromage, H.; Tempst, P.; Gauthier, N.P.; et al. TRIM3, a tumor suppressor linked to regulation of p21(Waf1/Cip1.). Oncogene 2014, 33, 308–315. [Google Scholar] [CrossRef]
- Song, Y.; Guo, Q.; Gao, S.; Hua, K. Tripartite motif-containing protein 3 plays a role of tumor inhibitor in cervical cancer. Biochem. Biophys. Res. Commun. 2018, 498, 686–692. [Google Scholar] [CrossRef]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar]
- Stramucci, L.; Pranteda, A.; Bossi, G. Insights of crosstalk between p53 protein and the MKK3/MKK6/p38 MAPK signaling pathway in cancer. Cancers 2018, 10, 131. [Google Scholar] [CrossRef]
- Micale, L.; Fusco, C.; Fontana, A.; Barbano, R.; Augello, B.; De Nittis, P.; Copetti, M.; Pellico, M.T.; Mandriani, B.; Cocciadiferro, D.; et al. TRIM8 downregulation in glioma affects cell proliferation and it is associated with patients survival. BMC Cancer 2015, 15, 470. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, B.; Shi, T.; Qin, H. MiR-182 promotes tumor growth and increases chemoresistance of human anaplastic thyroid cancer by targeting tripartite motif 8. OncoTargets Ther. 2017, 10, 1115–1122. [Google Scholar] [CrossRef]
- Caratozzolo, M.F.; Valletti, A.; Gigante, M.; Aiello, I.; Mastropasqua, F.; Marzano, F.; Ditonno, P.; Carrieri, G.; Simonnet, H.; D’Erchia, A.M.; et al. TRIM8 anti-proliferative action against chemo-resistant renal cell carcinoma. Oncotarget 2014, 5, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- Mastropasqua, F.; Marzano, F.; Valletti, A.; Aiello, I.; Di Tullio, G.; Morgano, A.; Liuni, S.; Ranieri, E.; Guerrini, L.; Gasparre, G.; et al. TRIM8 restores p53 tumour suppressor function by blunting N-MYC activity in chemo-resistant tumours. Mol. Cancer 2017, 16, 67. [Google Scholar] [CrossRef]
- Okumura, F.; Matsunaga, Y.; Katayama, Y.; Nakayama, K.I.; Hatakeyama, S. TRIM8 modulates STAT3 activity through negative regulation of PIAS3. J. Cell Sci. 2010, 123, 2238–2245. [Google Scholar] [CrossRef]
- Li, Q.; Yan, J.; Mao, A.P.; Li, C.; Ran, Y.; Shu, H.B.; Wang, Y.Y. Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 19341–19346. [Google Scholar] [CrossRef]
- Joo, H.M.; Kim, J.Y.; Jeong, J.B.; Seong, K.M.; Nam, S.Y.; Yang, K.H.; Kim, C.S.; Kim, H.S.; Jeong, M.; An, S.; et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur. J. Cell Biol. 2011, 90, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Liu, J.; Rao, J.; Lou, X.; Zhai, J.; Ni, Z.; Wang, X. Upregulated TRIM11 exerts its oncogenic effects in hepatocellular carcinoma through inhibition of P53. Cell Physiol. Biochem. 2017, 44, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.A.; van der Knaap, J.A.; Bot, A.G.M.; Mohd-Sarip, A.; Dekkers, D.H.W.; Timmermans, M.A.; Martens, J.W.M.; Demmers, J.A.A.; Verrijzer, C.P. Nucleotide biosynthetic enzyme GMP synthase is a TRIM21-controlled relay of p53 stabilization. Mol. Cell 2014, 53, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.Q.; Irby, R.B. TRIM21 is a novel regulator of Par-4 in colon and pancreatic cancer cells. Cancer Biol. Ther. 2017, 18, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of fatty acid Synthase by acetylation inhibits de novo lipogenesis and tumor cell growth. Cancer Res. 2016, 76, 6924–6936. [Google Scholar] [CrossRef]
- Jain, A.K.; Allton, K.; Duncan, A.D.; Barton, M.C. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol. Cell Biol. 2014, 34, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Yin, A.A.; Cheng, J.X.; Huang, H.Y.; Li, X.M.; Zhang, Y.Q.; Han, N.; Zhang, X. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene 2015, 34, 600–610. [Google Scholar] [CrossRef]
- Lv, D.; Li, Y.; Zhang, W.; Alvarez, A.A.; Song, L.; Tang, J.; Gao, W.-Q.; Hu, B.; Cheng, S.Y.; Feng, H. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat. Commun. 2017, 8, 1454. [Google Scholar] [CrossRef]
- Zhang, P.; Elabd, S.; Hammer, S.; Solozobova, V.; Yan, H.; Bartel, F.; Inoue, S.; Henrich, T.; Wittbrodt, J.; Loosli, F.; et al. TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene 2015, 34, 5729–5738. [Google Scholar] [CrossRef]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Lionnard, L.; Duc, P.; Brennan, M.S.; Kueh, A.J.; Pal, M.; Guardia, F.; Mojsa, B.; Damiano, M.-A.; Mora, S.; Lassot, I.; et al. TRIM17 and TRIM28 antagonistically regulate the ubiquitination and anti-apoptotic activity of BCL2A1. Cell Death Differ. 2018, 26, 902–917. [Google Scholar] [CrossRef]
- Sho, T.; Tsukiyama, T.; Sato, T.; Kondo, T.; Cheng, J.; Saku, T.; Asaka, M.; Hatakeyama, S. TRIM29 negatively regulates p53 via inhibition of Tip60. Biochim. Biophys Acta 2011, 1813, 1245–1253. [Google Scholar] [CrossRef]
- Guo, P.; Qiu, Y.; Ma, X.; Li, T.; Ma, X.; Zhu, L.; Lin, Y.; Han, L. Tripartite motif 31 promotes resistance to anoikis of hepatocarcinoma cells through regulation of p53–AMPK axis. Exp. Cell Res. 2018, 368, 59–66. [Google Scholar] [CrossRef]
- Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018, 8, 3224–3236. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef]
- Zhao, T.T.; Jin, F.; Li, J.G.; Xu, Y.Y.; Dong, H.T.; Liu, Q.; Xing, P.; Zhu, G.L.; Xu, H.; Yin, S.C.; et al. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-κB pathway. J. Cancer 2018, 9, 1349–1356. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, N.J.; Chen, C.; Tang, W.; Kornbluth, S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc. Natl. Acad. Sci. USA 2012, 109, 20931–20936. [Google Scholar] [CrossRef]
- Zhang, L.; Mei, Y.; Fu, N.; Guan, L.; Xie, W.; Liu, H.; Yu, C.; Yin, Z.; Yu, V.C.; You, H. TRIM39 regulates cell cycle progression and DNA damage responses via stabilizing p21. Proc. Natl. Acad. Sci. USA 2012, 109, 20937–20942. [Google Scholar] [CrossRef]
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.Q. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 2014, 147, 1043–1054. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.; Zhao, L.; Su, L.; Diao, K.; Mi, X. TRIM59 overexpression correlates with poor prognosis and contributes to breast cancer progression through AKT signaling pathway. Mol. Carcinog. 2018, 57, 1792–1802. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Yang, H.; Shi, G.; Xu, G.; Shi, J.; Yin, N.; Chen, D. TRIM66 overexpresssion contributes to osteosarcoma carcinogenesis and indicates poor survival outcome. Oncotarget 2015, 6, 23708–23719. [Google Scholar] [CrossRef]
- Tan, Z.; Song, L.; Wu, W.; Zhou, Y.; Zhu, J.; Wu, G.; Cao, L.; Song, J.; Li, J.; Zhang, W. TRIM14 promotes chemoresistance in gliomas by activating Wnt/β-catenin signaling via stabilizing Dvl2. Oncogene 2018, 37, 5403–5415. [Google Scholar] [CrossRef]
- Wu, G.; Song, L.; Zhu, J.; Hu, Y.; Cao, L.; Tan, Z.; Zhang, S.; Li, Z.; Li, J. An ATM/TRIM37/NEMO Axis Counteracts Genotoxicity by Activating Nuclear-to-Cytoplasmic NF-κB Signaling. Cancer Res. 2018, 78, 6399–6412. [Google Scholar] [CrossRef]
- Noguchi, K.; Okumura, F.; Takahashi, N.; Kataoka, A.; Kamiyama, T.; Todo, S.; Hatakeyama, S. TRIM40 promotes neddylation of IKKγ and is downregulated in gastrointestinal cancers. Carcinogenesis 2011, 32, 995–1004. [Google Scholar] [CrossRef]
- Chen, L.; Yang, X. TRIM11 cooperates with HSF1 to suppress the anti-tumor effect of proteotoxic stress drugs. Cell Cycle 2019, 18, 60–68. [Google Scholar] [CrossRef]
- Miao, Z.F.; Wang, Z.N.; Zhao, T.T.; Xu, Y.Y.; Wu, J.H.; Liu, X.Y.; Xu, H.; You, Y.; Xu, H.M. TRIM24 is upregulated in human gastric cancer and promotes gastric cancer cell growth and chemoresistance. Virchows Arch. Int. J. Pathol. 2015, 466, 525–532. [Google Scholar] [CrossRef]
- Damineni, S.; Balaji, S.A.; Shettar, A.; Nayanala, S.; Kumar, N.; Kruthika, B.S.; Subramanian, K.; Vijayakumar, M.; Mukherjee, G.; Gupta, V.; et al. Expression of tripartite motif-containing protein 28 in primary breast carcinoma predicts metastasis and is involved in the stemness, chemoresistance, and tumor growth. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39. [Google Scholar] [CrossRef]
- Yang, H.; Palmbos, P.L.; Wang, L.; Kim, E.H.; Ney, G.M.; Liu, C.; Prasad, J.; Misek, D.E.; Yu, X.; Ljungman, M.; et al. ATDC (Ataxia Telangiectasia Group D Complementing) promotes radioresistance through an interaction with the RNF8 ubiquitin ligase. J. Biol. Chem. 2015, 290, 27146–27157. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, W.; Du, B.; Zang, S.; Wang, X.; Mao, X.; Hu, Z. TRIM32 overexpression improves chemoresistance through regulation of mitochondrial function in non-small-cell lung cancers. OncoTargets Ther. 2018, 11, 7841–7852. [Google Scholar] [CrossRef]
- Fan, W.; Du, F.; Liu, X. TRIM66 confers tumorigenicity of hepatocellular carcinoma cells by regulating GSK-3β-dependent Wnt/β-catenin signaling. Eur. J. Pharmacol. 2019, 850, 109–117. [Google Scholar] [CrossRef]
- Wang, L.; Shang, X.; Feng, Q. LncRNA TATDN1 contributes to the cisplatin resistance of non-small cell lung cancer through TATDN1/miR-451/TRIM66 axis. Cancer Biol. Ther. 2019, 20, 261–271. [Google Scholar] [CrossRef]
- Wang, X.; Guo, H.; Yao, B.; Helms, J. MiR-15b inhibits cancer-initiating cell phenotypes and chemoresistance of cisplatin by targeting TRIM14 in oral tongue squamous cell cancer. Oncol. Rep. 2017, 37, 2720–2726. [Google Scholar] [CrossRef]
- Elabd, S.; Meroni, G.; Blattner, C. TRIMming p53’s anticancer activity. Oncogene 2016, 35, 5577–5584. [Google Scholar] [CrossRef] [PubMed]
- Gatt, M.E.; Takada, K.; Mani, M.; Lerner, M.; Pick, M.; Hideshima, T.; Carrasco, D.E.; Protopopov, A.; Ivanova, E.; Sangfelt, O.; et al. TRIM13 (RFP2) downregulation decreases tumour cell growth in multiple myeloma through inhibition of NF κB pathway and proteasome activity. Br. J. Haematol. 2013, 162, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Kasamatsu, A.; Endo-Sakamoto, Y.; Eizuka, K.; Hiroshima, K.; Kita, A.; Saito, T.; Koike, K.; Tanzawa, H.; Uzawa, K. Increased expression of tripartite motif (TRIM) like 2 promotes tumoral growth in human oral cancer. Biochem. Biophys. Res Commun. 2019, 508, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; He, D.; He, K.; Zhang, Q.; Tang, M.; Dai, J.; Lv, H.; Wang, X.; Xiang, G.; Yu, H. Downregulation of TRIM21 contributes to hepatocellular carcinoma carcinogenesis and indicates poor prognosis of cancers. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 8761–8772. [Google Scholar] [CrossRef]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef]
- Lee, D.S.; Grandis, J.R.; Johnson, D.E. Chapter 7—STAT3 as a Major Contributor to Chemoresistance. In Targeting Cell Survival Pathways to Enhance Response to Chemotherapy, 1st ed.; Johnson, D.E., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 145–167. [Google Scholar]
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 2002, 277, 37315–37322. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H.; Li, Z.; Zhao, Z.; Yip-Schneider, M.; Fan, Q.; Schmidt, C.M.; Chiorean, E.G.; Xie, J.; Cheng, L.; et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int. J. Biochem. Mol. Biol. 2011, 2, 89–98. [Google Scholar]
- Tian, S.; Li, P.; Sheng, S.; Jin, X. Upregulation of pyruvate kinase M2 expression by fatty acid synthase contributes to gemcitabine resistance in pancreatic cancer. Oncol. Lett. 2018, 15, 2211–2217. [Google Scholar] [CrossRef]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef]
- Bader, A.G.; Lammers, P. The Therapeutic Potential of microRNAs. Available online: www.iptonline.com/articles/public/TheTherapeuticPotentialofmicroRNAs.pdf (accessed on 2 April 2019).
- Shen, M.; Chan, T.H.; Dou, Q.P. Targeting tumor ubiquitin-proteasome pathway with polyphenols for chemosensitization. Anticancer Agents Med. Chem. 2012, 12, 891–901. [Google Scholar] [CrossRef]
- Weissman, A.M.; Yang, Y.; Kitagaki, J.; Sasiela, C.A.; Beutler, J.A.; O’Keefe, B.R. Inhibiting Hdm2 and ubiquitin-activating enzyme: Targeting the ubiquitin conjugating system in cancer. Ernst Schering Res. Found. Symp. Proc. 2008, 1, 171–190. [Google Scholar]
- Clement, J.A.; Kitagaki, J.; Yang, Y.; Saucedo, C.J.; O’Keefe, B.R.; Weissman, A.M.; McKee, T.C.; McMahon, J.B. Discovery of new pyridoacridine alkaloids from Lissoclinum cf. badium that inhibit the ubiquitin ligase activity of Hdm2 and stabilize p53. Bioorg. Med. Chem. 2008, 16, 10022–10028. [Google Scholar] [CrossRef]
- Sasiela, C.A.; Stewart, D.H.; Kitagaki, J.; Safiran, Y.J.; Yang, Y.; Weissman, A.M.; Oberoi, P.; Davydov, I.V.; Goncharova, E.; Beutler, J.A.; et al. Identification of inhibitors for MDM2 ubiquitin ligase activity from natural product extracts by a novel high-throughput electrochemiluminescent screen. J. Biomol. Screen. 2008, 13, 229–237. [Google Scholar] [CrossRef]
- Vinod, B.S.; Maliekal, T.T.; Anto, R.J. Phytochemicals as chemosensitizers: From molecular mechanism to clinical significance. Antioxid. Redox Signal. 2013, 18, 1307–1348. [Google Scholar] [CrossRef]
- Rezaee, R.; Momtazi, A.A.; Monemi, A.; Sahebkar, A. Curcumin: A potentially powerful tool to reverse cisplatin-induced toxicity. Pharmacol. Res. 2017, 117, 218–227. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Chong, T.; Yang, J.; Li, H.; Chen, H. Curcumin enhances the radiosensitivity of renal cancer cells by suppressing NF-κB signaling pathway. Biomed. Pharmacother. 2017, 94, 974–981. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.; Fan, Y.; Guan, B.; Jia, J.; Gao, Y.; Wang, K.; Wu, K.; Wang, X.; Zheng, P.; et al. Curcumin enhances temsirolimus-induced apoptosis in human renal carcinoma cells through upregulation of YAP/p53. Oncol. Lett. 2016, 12, 4999–5006. [Google Scholar] [CrossRef]
- Saydmohammed, M.; Joseph, D.; Syed, V. Curcumin suppresses constitutive activation of STAT-3 by up-regulating protein inhibitor of activated STAT-3 (PIAS-3) in ovarian and endometrial cancer cells. J. Cell Biochem. 2010, 110, 447–456. [Google Scholar] [CrossRef]
- Moustakim, M.; Clark, P.G.K.; Hay, D.A.; Dixon, D.J.; Brennan, P.E. Chemical probes and inhibitors of bromodomains outside the BET family. MedChemComm 2016, 7, 2246–2264. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, G.P.; Zhao, F.; Li, R.Q.; Liu, S.J.; Zhao, Z.R.; Wang, X. Target therapy of TRIM-14 inhibits osteosarcoma aggressiveness through the nuclear factor-κB signaling pathway. Exp. Ther. Med. 2018, 15, 2365–2373. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| TRIM Proteins and Chemoresistance Pathways | Ref. | |
|---|---|---|
| p53 Positive Regulators | ||
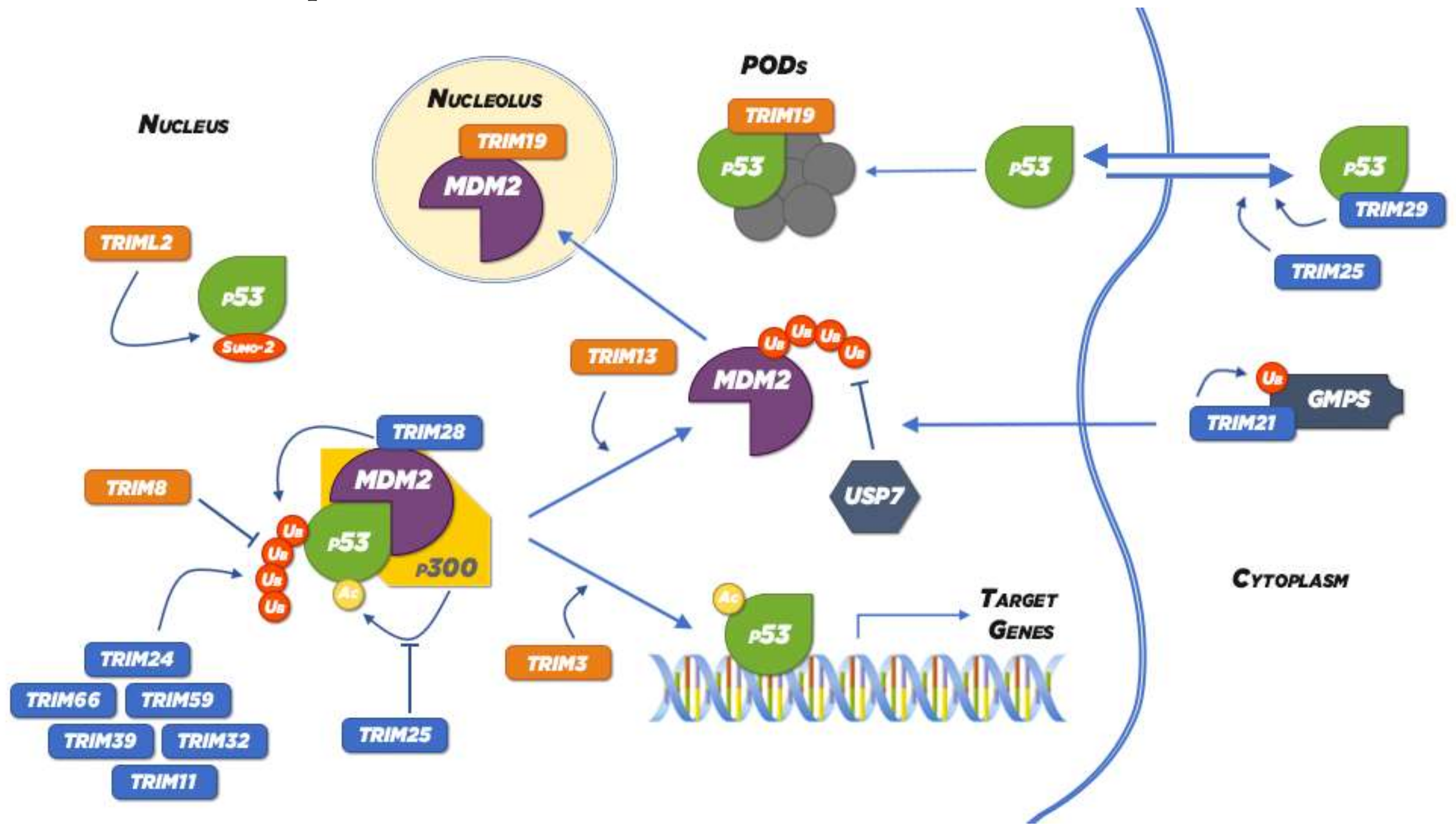
| TRIML2 * | p53 sumoylation | [47] |
| TRIM3 * | p53 stabilization; p21 sequestration (preventing cyclin D1-cdk4 accumulation); p38 signalling pathway inactivation | [48,49,50,51,52] |
| TRIM8 * | Impairment of the interaction between p53 and MDM2; PIAS3 ubiquitylation (activation of NF-κB and STAT3 pathways); TAK1 activation (enhancement of the NF-κB pathway) | [53,54,55,56,57,58] |
| TRIM13 | MDM2 polyubiquitylation and proteasomal degradation | [59] |
| TRIM19 * | Recruits p53 into the PML-NBs; Sequestrates MDM2 into the nucleolus | [60] |
| p53 Negative Regulators | ||
| TRIM11 | p53 down-regulation | [61] |
| TRIM21 | GMPS ubiquitylation and sequestration into the cytoplasm; PAR-4 down-regulates PAR-4; FASN ubiquitylation for degradation | [62,63,64] |
| TRIM24 * | p53 ubiquitylation for degradation; Induction of the expression of PI3KCA (activation of PI3K/Akt and NF-κB pathways); Co-transcriptional activator (recruitment of STAT3) | [65,66,67] |
| TRIM25 | Interferes with the formation of the complex p53-MDM2-p300; Relocalization of p53 into the cytoplasm by interacting with G3BP2 | [68,69] |
| TRIM28 | Interaction with MDM2 for targeting p53 for proteasomal degradation; Interaction (inhibited by TRIM17) with the anti-apoptotic BCL2A1 to induce its ubiquitylation and degradation | [70,71] |
| TRIM29 | Sequestration of p53 into the cytoplasm; Degradation of Tip60 (inhibition of p53 acetylation); Binding to the DNA repair factor RNF8 | [72] |
| TRIM31 | K48-linked polyubiquitylation and proteasomal degradation of p53; polyubiquitylation of TRAF2 upregulating the levels of nuclear p65 (NF-κB) | [73,74] |
| TRIM32 * | Degradation of p53; Upregulation of the phosphorylation of IkB | [75,76] |
| TRIM39 | p53 ubiquitylation for degradation; p21 stabilization (by preventing its interaction with Cdt2) | [77,78] |
| TRIM59 | Enhancement of p53 ubiquitylation and proteasomal degradation; Reduction of caspases activation; Upregulation of Bcl-2 and Bcl-xL, increasing Akt phosphorylation | [79,80] |
| TRIM66 | Down-regulation of p53 and caspases 7 and 9 | [81] |
| Other Mechanisms | ||
| TRIM14 | Dvl2 binding and stabilization (activation of Wnt-beta catenin pathway and the expression of MGMT) | [82] |
| TRIM37 | Nuclear export of NEMO (IKK/NF-κB activation) | [83] |
| TRIM40 | Neddylation of IKKγ (inhibition of NF-κB-mediated cell growth) | [84] |
| TRIM-Mediated Chemoresistance in Cancers | ||||
|---|---|---|---|---|
| TRIM Proteins | Expression | Cancer | Chemotherapeutic Drug | References |
| TRIM8 | ↓ | ccRCC-TS and XE | nutlin-3, cisplatin, axitinib and sorafenib | [55,56] |
| TRIM11 | ↑ | HCT116 and HEK293T | proteasome inhibitor bortezomib (BTZ)16, autophagy inhibitor chloroquine (CQ)17, piperlongumine (PL) and celastrol | [85] |
| TRIM24 | ↑ | GC-TS | 5-fluorouracil | [86] |
| TRIM28 | ↑ | BC-TS, MDA-MB-231 and BT-474 | doxorubicin, 5-fluorouracil, and methotrexate | [87] |
| TRIM29 | ↑ | HEK293, Panc1, BxPc3 and CAPAN2 | cytotoxic chemotherapy and ionizing radiation | [88] |
| TRIM31 | ↑ | PC-TS and HPDECs | gemcitabine | [74] |
| TRIM32 | ↑ | BC-TS, MCF-7, T-47D / NSCLC-TS, HBE, A549, H1299, H460, H358, H3255, H1975, H2228 | cisplatin | [76,89] |
| TRIM39 | ↑ | hTERT-RPE | nutlin-3a | [77] |
| TRIM59 | ↑ | BC-TS, MCF-7 and SK-BR-3 | paclitaxel | [80] |
| TRIM66 | ↑ | NSCLC-TS, Hep-3B, SNU-449 and HL-7702 | cisplatin | [90,91] |
| TRIM14 | ↑ | OTSC-TS and SCC25 | cisplatin | [92] |
| TRIM37 | ↑ | EC-TS and PCL, Eca109 | cisplatin | [83] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valletti, A.; Marzano, F.; Pesole, G.; Sbisà, E.; Tullo, A. Targeting Chemoresistant Tumors: Could TRIM Proteins-p53 Axis Be a Possible Answer? Int. J. Mol. Sci. 2019, 20, 1776. https://doi.org/10.3390/ijms20071776
Valletti A, Marzano F, Pesole G, Sbisà E, Tullo A. Targeting Chemoresistant Tumors: Could TRIM Proteins-p53 Axis Be a Possible Answer? International Journal of Molecular Sciences. 2019; 20(7):1776. https://doi.org/10.3390/ijms20071776
Chicago/Turabian StyleValletti, Alessio, Flaviana Marzano, Graziano Pesole, Elisabetta Sbisà, and Apollonia Tullo. 2019. "Targeting Chemoresistant Tumors: Could TRIM Proteins-p53 Axis Be a Possible Answer?" International Journal of Molecular Sciences 20, no. 7: 1776. https://doi.org/10.3390/ijms20071776
APA StyleValletti, A., Marzano, F., Pesole, G., Sbisà, E., & Tullo, A. (2019). Targeting Chemoresistant Tumors: Could TRIM Proteins-p53 Axis Be a Possible Answer? International Journal of Molecular Sciences, 20(7), 1776. https://doi.org/10.3390/ijms20071776