Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease

Abstract
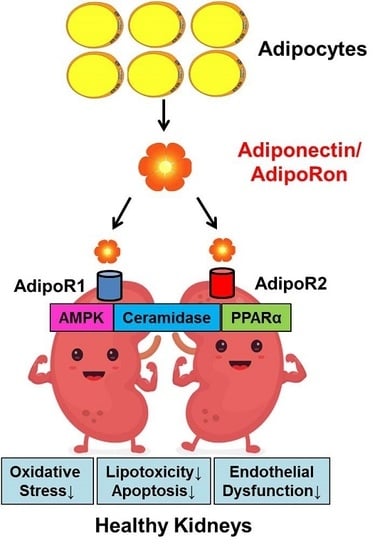
:
{kind=link}
{kind=link}
1. Introduction
2. Adiponectin in Renal Physiology: Its Association with Albuminuria and Glomerular Filtration Rate
3. Expression of Adiponectin and Its Receptors and Their Implication for Renoprotection
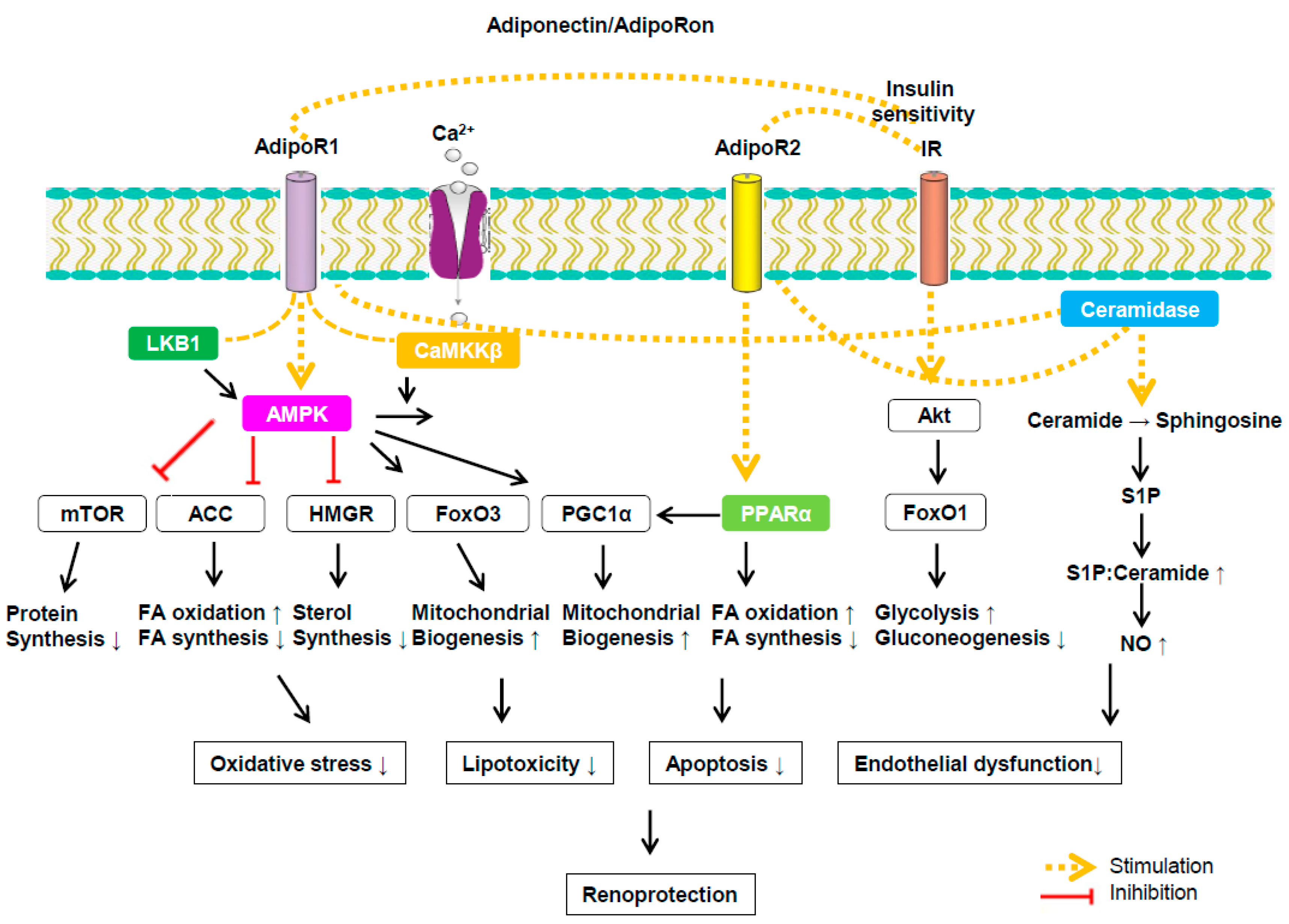
4. Signaling Pathways Associated with Adiponectin and Its Receptor Binding
5. Adiponectin and Adiponectin Receptors as Therapeutic Targets for DKD
5.1. Strategy for Upregulation of Adiponectin and Its Receptors
5.2. Development of an AdipoR Agonist, AdipoRon
5.3. Role of AdipoRon in DKD
6. Conclusions and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Park, C.W. Diabetic kidney disease: From epidemiology to clinical perspectives. Diabetes Metab. J. 2014, 38, 252–260. [Google Scholar] [CrossRef]
- Martinez de Morentin, P.B.; Varela, L.; Ferno, J.; Nogueiras, R.; Dieguez, C.; Lopez, M. Hypothalamic lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 350–361. [Google Scholar] [CrossRef]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Martinez Cantarin, M.P.; Waldman, S.A.; Doria, C.; Frank, A.M.; Maley, W.R.; Ramirez, C.B.; Keith, S.W.; Falkner, B. The adipose tissue production of adiponectin is increased in end-stage renal disease. Kidney Int. 2013, 83, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bao, B.J.; Fan, Y.P.; Shi, L.; Li, S.Q. Changes of adiponectin and its receptors in rats following chronic renal failure. Ren. Fail. 2014, 36, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Esmaili, S.; Xu, A.; George, J. The multifaceted and controversial immunometabolic actions of adiponectin. TEM 2014, 25, 444–451. [Google Scholar] [CrossRef]
- Halberg, N.; Schraw, T.D.; Wang, Z.V.; Kim, J.Y.; Yi, J.; Hamilton, M.P.; Luby-Phelps, K.; Scherer, P.E. Systemic fate of the adipocyte-derived factor adiponectin. Diabetes 2009, 58, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Shimotomai, T.; Kakei, M.; Narita, T.; Koshimura, J.; Hosoba, M.; Kato, M.; Komatsuda, A.; Ito, S. Enhanced urinary adiponectin excretion in IgA-nephropathy patients with proteinuria. Ren. Fail. 2005, 27, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Y.; Hughes, J.T.; Charlesworth, J.A.; Kelly, J.J.; Peake, P.W. Adiponectin is present in the urine in its native conformation, and specifically reduces the secretion of MCP-1 by proximal tubular cells. Nephrology 2008, 13, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Barlovic, D.P.; Zaletel, J.; Prezelj, J. Adipocytokines are associated with renal function in patients with normal range glomerular filtration rate and type 2 diabetes. Cytokine 2009, 46, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Barlovic, D.P.; Zaletel, J.; Prezelj, J. Association between adiponectin and low-grade albuminuria is BMI-dependent in type 2 diabetes. Kidney Blood Press. Res. 2010, 33, 405–410. [Google Scholar] [CrossRef]
- Tamba, S.; Nakatsuji, H.; Kishida, K.; Noguchi, M.; Ogawa, T.; Okauchi, Y.; Nishizawa, H.; Imagawa, A.; Nakamura, T.; Matsuzawa, Y.; et al. Relationship between visceral fat accumulation and urinary albumin-creatinine ratio in middle-aged Japanese men. Atherosclerosis 2010, 211, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Jorsal, A.; Petersen, E.H.; Tarnow, L.; Hess, G.; Zdunek, D.; Frystyk, J.; Flyvbjerg, A.; Lajer, M.; Rossing, P. Urinary adiponectin excretion rises with increasing albuminuria in type 1 diabetes. J. Diabetes Complicat. 2013, 27, 604–608. [Google Scholar] [CrossRef]
- Kacso, I.M.; Bondor, C.I.; Kacso, G. Plasma adiponectin is related to the progression of kidney disease in type 2 diabetes patients. Scand. J. Clin. Lab. Investig. 2012, 72, 333–339. [Google Scholar] [CrossRef]
- Bjornstad, P.; Pyle, L.; Kinney, G.L.; Rewers, M.; Johnson, R.J.; Maahs, D.M.; Snell-Bergeon, J.K. Adiponectin is associated with early diabetic kidney disease in adults with type 1 diabetes: A Coronary Artery Calcification in Type 1 Diabetes (CACTI) Study. J. Diabetes Complicat. 2017, 31, 369–374. [Google Scholar] [CrossRef]
- Lim, C.C.; Teo, B.W.; Tai, E.S.; Lim, S.C.; Chan, C.M.; Sethi, S.; Wong, T.Y.; Sabanayagam, C. Elevated serum leptin, adiponectin and leptin to adiponectin ratio is associated with chronic kidney disease in Asian adults. PLoS ONE 2015, 10, e0122009. [Google Scholar] [CrossRef]
- Park, J.T.; Yoo, T.H.; Kim, J.K.; Oh, H.J.; Kim, S.J.; Yoo, D.E.; Lee, M.J.; Shin, D.H.; Han, S.H.; Han, D.S.; et al. Leptin/adiponectin ratio is an independent predictor of mortality in nondiabetic peritoneal dialysis patients. Perit. Dial. Int. 2013, 33, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Von Eynatten, M.; Liu, D.; Hock, C.; Oikonomou, D.; Baumann, M.; Allolio, B.; Korosoglou, G.; Morcos, M.; Campean, V.; Amann, K.; et al. Urinary adiponectin excretion: A novel marker for vascular damage in type 2 diabetes. Diabetes 2009, 58, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Perri, A.; Vizza, D.; Lofaro, D.; Gigliotti, P.; Leone, F.; Brunelli, E.; Malivindi, R.; De Amicis, F.; Romeo, F.; De Stefano, R.; et al. Adiponectin is expressed and secreted by renal tubular epithelial cells. J. Nephrol. 2013, 26, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ramachandrarao, S.; Qiu, G.; Usui, H.K.; Zhu, Y.; Dunn, S.R.; Ouedraogo, R.; Hough, K.; McCue, P.; Chan, L.; et al. Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Investig. 2008, 118, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, J.M.; Wang, Z.V.; Park, A.S.; Zhang, J.; Zhang, D.; Hu, M.C.; Moe, O.W.; Susztak, K.; Scherer, P.E. Adiponectin promotes functional recovery after podocyte ablation. JASN 2013, 24, 268–282. [Google Scholar] [CrossRef]
- Fang, F.; Liu, G.C.; Kim, C.; Yassa, R.; Zhou, J.; Scholey, J.W. Adiponectin attenuates angiotensin II-induced oxidative stress in renal tubular cells through AMPK and cAMP-Epac signal transduction pathways. Am. J. Physiol. Ren. Physiol. 2013, 304, F1366–F1374. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. JASN 2018, 29, 1108–1127. [Google Scholar] [CrossRef]
- Choi, S.R.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Kim, Y.; Choi, B.S.; Kim, Y.S.; Kim, H.W.; Lim, K.M.; Kim, M.J.; et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metab. Clin. Exp. 2018, 85, 348–360. [Google Scholar] [CrossRef]
- Tsuchida, A.; Yamauchi, T.; Ito, Y.; Hada, Y.; Maki, T.; Takekawa, S.; Kamon, J.; Kobayashi, M.; Suzuki, R.; Hara, K.; et al. Insulin/Foxo1 pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. J. Biol. Chem. 2004, 279, 30817–30822. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Hug, C.; Wang, J.; Ahmad, N.S.; Bogan, J.S.; Tsao, T.S.; Lodish, H.F. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. USA 2004, 101, 10308–10313. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Mao, X.; Kikani, C.K.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.Y.; et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.W. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res. Clin. Pract. 2016, 35, 69–77. [Google Scholar] [CrossRef]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef]
- Jensen, T.E.; Sylow, L.; Rose, A.J.; Madsen, A.B.; Angin, Y.; Maarbjerg, S.J.; Richter, E.A. Contraction-stimulated glucose transport in muscle is controlled by AMPK and mechanical stress but not sarcoplasmatic reticulum Ca2+ release. Mol. Metab. 2014, 3, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Fioretto, P.; Mauer, M. Histopathology of diabetic nephropathy. Semin. Nephrol. 2007, 27, 195–207. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.W. New therapeutic agents in diabetic nephropathy. Korean J. Intern. Med. 2017, 32, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.M.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef]
- Chiribau, C.B.; Cheng, L.; Cucoranu, I.C.; Yu, Y.S.; Clempus, R.E.; Sorescu, D. FOXO3A regulates peroxiredoxin III expression in human cardiac fibroblasts. J. Biol. Chem. 2008, 283, 8211–8217. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Ko, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1alpha axis in db/db mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, H.W.; Kim, T.Y.; et al. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 28, 1543–1561. [Google Scholar] [CrossRef]
- Luo, X.; Deng, L.; Lamsal, L.P.; Xu, W.; Xiang, C.; Cheng, L. AMP-activated protein kinase alleviates extracellular matrix accumulation in high glucose-induced renal fibroblasts through mTOR signaling pathway. Cell. Physiol. Biochem. 2015, 35, 191–200. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Kim, Y.; Hwang, S.D.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Choi, B.S.; Kim, Y.S.; Kim, H.W.; Park, C.W. Attenuated Lymphatic Proliferation Ameliorates Diabetic Nephropathy and High-Fat Diet-Induced Renal Lipotoxicity. Sci. Rep. 2019, 9, 1994. [Google Scholar] [CrossRef]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef]
- Kuzmenko, D.I.; Klimentyeva, T.K. Role of Ceramide in Apoptosis and Development of Insulin Resistance. Biochem. Biokhimiia 2016, 81, 913–927. [Google Scholar] [CrossRef]
- Summers, S.A. The ART of Lowering Ceramides. Cell Metab. 2015, 22, 195–196. [Google Scholar] [CrossRef] [Green Version]
- Symons, J.D.; Abel, E.D. Lipotoxicity contributes to endothelial dysfunction: A focus on the contribution from ceramide. Rev. Endocr. Metab. Dis. 2013, 14, 59–68. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Deng, X.; Ito, T.; Carr, B.K.; May, W.S. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J. Biol. Chem. 1999, 274, 20296–20300. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Holland, W.L.; Wilson, L.; Tanner, J.M.; Kearns, D.; Cahoon, J.M.; Pettey, D.; Losee, J.; Duncan, B.; Gale, D.; et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012, 61, 1848–1859. [Google Scholar] [CrossRef]
- Sugiyama, S.; Fukushima, H.; Kugiyama, K.; Maruyoshi, H.; Kojima, S.; Funahashi, T.; Sakamoto, T.; Horibata, Y.; Watanabe, K.; Koga, H.; et al. Pravastatin improved glucose metabolism associated with increasing plasma adiponectin in patients with impaired glucose tolerance and coronary artery disease. Atherosclerosis 2007, 194, e43–e51. [Google Scholar] [CrossRef]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Yamaguchi, F.; Ishihara, Y.; Ishihara, N.; Noma, T.; Tokuda, M.; Kohno, M. Valsartan ameliorates the constitutive adipokine expression pattern in mature adipocytes: A role for inverse agonism of the angiotensin II type 1 receptor in obesity. Hypertens. Res. 2014, 37, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Unger, T. Vascular protection in diabetes: A pharmacological view of angiotensin II type 1 receptor blockers. Acta Diabetol. 2005, 42, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, D.; Li, J.; Zhang, X.; Fan, F.; Guan, Y. Role of PPARgamma in renoprotection in Type 2 diabetes: Molecular mechanisms and therapeutic potential. Clin. Sci. 2009, 116, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, F.; Asztalos, I.B.; Horvath, K.V.; Steiner, G.; Schaefer, E.J.; Asztalos, B.F. Fenofibrate, HDL, and cardiovascular disease in Type-2 diabetes: The DAIS trial. Atherosclerosis 2016, 247, 35–39. [Google Scholar] [CrossRef]
- Alderete, T.L.; Sattler, F.R.; Richey, J.M.; Allayee, H.; Mittelman, S.D.; Sheng, X.; Tucci, J.; Gyllenhammer, L.E.; Grant, E.G.; Goran, M.I. Salsalate treatment improves glycemia without altering adipose tissue in nondiabetic obese hispanics. Obesity 2015, 23, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Matsuda, M.; Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Makishima, M.; Shimomura, I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 2003, 52, 1655–1663. [Google Scholar] [CrossRef]
- Yu, J.G.; Javorschi, S.; Hevener, A.L.; Kruszynska, Y.T.; Norman, R.A.; Sinha, M.; Olefsky, J.M. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes 2002, 51, 2968–2974. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Cersosimo, E.; Triplitt, C.; DeFronzo, R.A. Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 2007, 72, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Guarente, L. Sirtuins as potential targets for metabolic syndrome. Nature 2006, 444, 868–874. [Google Scholar] [CrossRef]
- Wei, W.; Dutchak, P.A.; Wang, X.; Ding, X.; Wang, X.; Bookout, A.L.; Goetz, R.; Mohammadi, M.; Gerard, R.D.; Dechow, P.C.; et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proc. Natl. Acad. Sci. USA 2012, 109, 3143–3148. [Google Scholar] [CrossRef] [PubMed]
- Ayerden Ebinc, F.; Ebinc, H.; Derici, U.; Aral, A.; Aybay, C.; Tacoy, G.; Koc, E.; Mutluay, R.; Altok Reis, K.; Erten, Y.; et al. The relationship between adiponectin levels and proinflammatory cytokines and left ventricular mass in dialysis patients. J. Nephrol. 2009, 22, 216–223. [Google Scholar]
- Durand, J.L.; Nawrocki, A.R.; Scherer, P.E.; Jelicks, L.A. Gender differences in adiponectin modulation of cardiac remodeling in mice deficient in endothelial nitric oxide synthase. J. Cell. Biochem. 2012, 113, 3276–3287. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Combs, T.P.; Pajvani, U.B.; Berg, A.H.; Lin, Y.; Jelicks, L.A.; Laplante, M.; Nawrocki, A.R.; Rajala, M.W.; Parlow, A.F.; Cheeseboro, L.; et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 2004, 145, 367–383. [Google Scholar] [CrossRef]
- Reid, I.R.; Cornish, J.; Baldock, P.A. Nutrition-related peptides and bone homeostasis. J. Bone Miner. Res. 2006, 21, 495–500. [Google Scholar] [CrossRef]
- Ealey, K.N.; Kaludjerovic, J.; Archer, M.C.; Ward, W.E. Adiponectin is a negative regulator of bone mineral and bone strength in growing mice. Exp. Biol. Med. 2008, 233, 1546–1553. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Iwabu, M.; Ueki, K.; Yamauchi, T.; Kadowaki, T. Perspective of Small-Molecule AdipoR Agonist for Type 2 Diabetes and Short Life in Obesity. Diabetes Metab. J. 2015, 39, 363–372. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Park, C.W. Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1782. https://doi.org/10.3390/ijms20071782
Kim Y, Park CW. Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease. International Journal of Molecular Sciences. 2019; 20(7):1782. https://doi.org/10.3390/ijms20071782
Chicago/Turabian StyleKim, Yaeni, and Cheol Whee Park. 2019. "Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease" International Journal of Molecular Sciences 20, no. 7: 1782. https://doi.org/10.3390/ijms20071782
APA StyleKim, Y., & Park, C. W. (2019). Mechanisms of Adiponectin Action: Implication of Adiponectin Receptor Agonism in Diabetic Kidney Disease. International Journal of Molecular Sciences, 20(7), 1782. https://doi.org/10.3390/ijms20071782