Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System

 ,
,  ,
,
Abstract
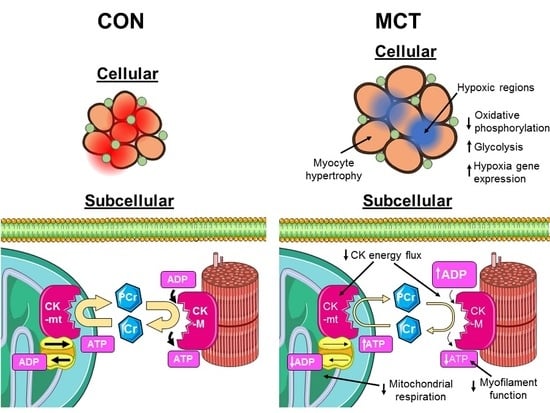
:
1. Introduction
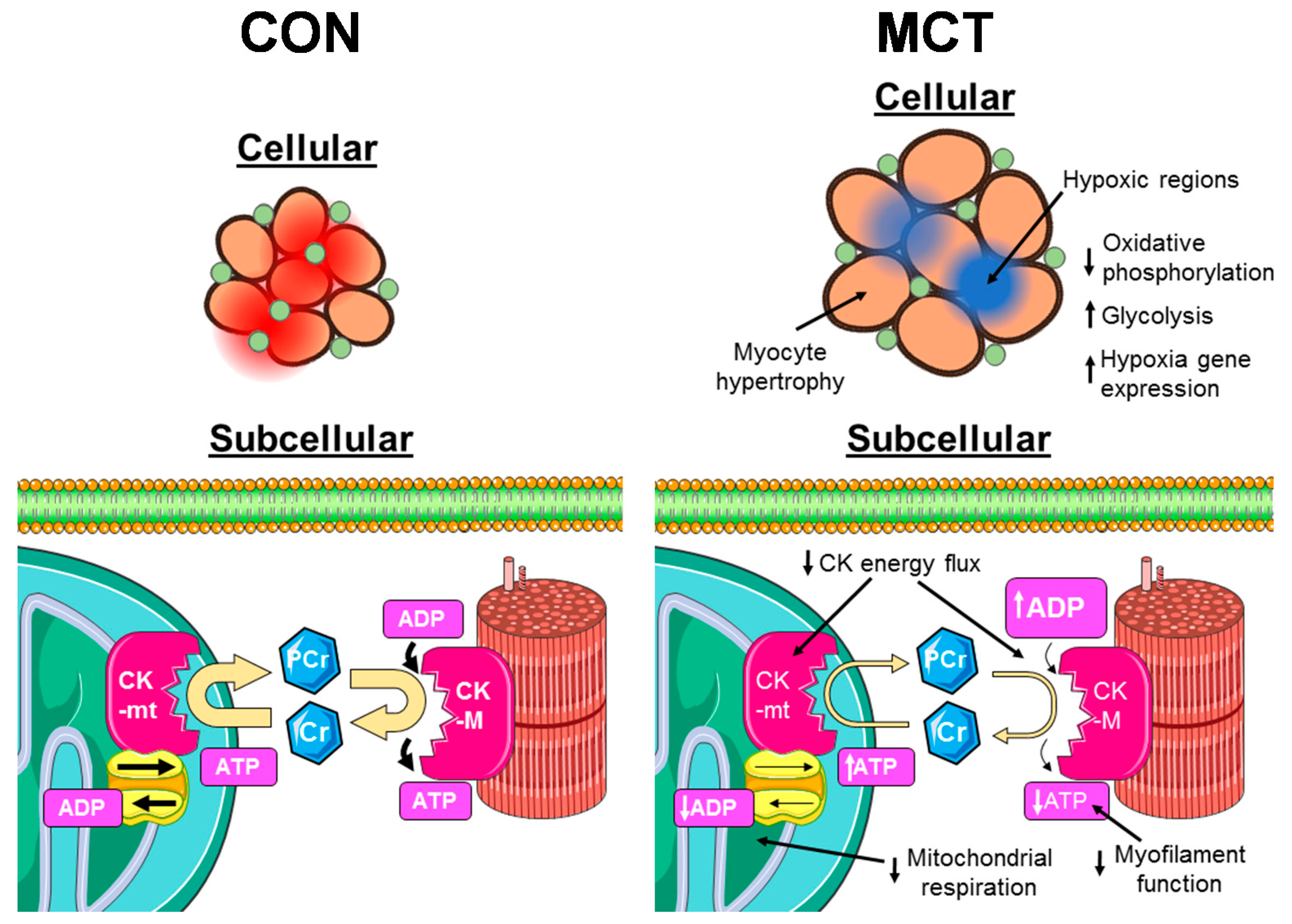
2. Results
3. Discussion
3.1. Insufficient Capillary Supply Produces Extensive Myocardial Hypoxia in Monocrotaline Rats
3.2. Reduced Mitochondrial Synthesis of Phosphocreatine in Monocrotaline Rats
3.3. Limitations
3.4. Conclusion
4. Materials and Methods
4.1. Histology
4.2. Computer Modelling O2 Distribution
4.3. Mitochondrial Respiration in Isolated Myocytes
4.4. Enzyme Activity Assays
4.5. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BB | beta-adrenergic receptor blocker |
| CK | creatine kinase |
| CON | saline-injected control |
| Cr | creatine |
| MCT | monocrotaline |
| MCT+BB | monocrotaline + beta blocker |
| PCr | phosphocreatine |
| SERCA | sarco(endo)plasmic reticulum Ca2+ ATPase |
References
- Haddad, F.; Doyle, R.; Murphy, D.J.; Hunt, S.A. Right Ventricular Function in Cardiovascular Disease, Part II: Pathophysiology, Clinical Importance, and Management of Right Ventricular Failure. Circulation 2008, 117, 1717–1731. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Vonk Noordegraaf, A.; Westerhof, B.E.; Westerhof, N. The Relationship Between the Right Ventricle and its Load in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2017, 69, 236–243. [Google Scholar] [CrossRef]
- Henkens, I.R.; Van Wolferen, S.A.; Gan, C.T.-J.; Boonstra, A.; Swenne, C.A.; Twisk, J.W.; Kamp, O.; van der Wall, E.E.; Schalij, M.J.; Vonk-Noordegraaf, A.; et al. Relation of Resting Heart Rate to Prognosis in Patients with Idiopathic Pulmonary Arterial Hypertension. Am. J. Cardiol. 2009, 103, 1451–1456. [Google Scholar] [CrossRef]
- Wong, Y.Y.; Westerhof, N.; Ruiter, G.; Lubberink, M.; Raijmakers, P.; Knaapen, P.; Marcus, J.T.; Boonstra, A.; Lammertsma, A.A.; van der Laarse, W.J.; et al. Systolic pulmonary artery pressure and heart rate are main determinants of oxygen consumption in the right ventricular myocardium of patients with idiopathic pulmonary arterial hypertension. Eur. J. Heart Fail. 2011, 13, 1290–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfarth, T.; Gerbershagen, H.-P.; Giessler, C.; Leineweber, K.; Heinroth-Hoffmann, I.; Pönicke, K.; Brodde, O.-E. The Cardiac β -Adrenoceptor-G-protein(s)-adenylyl Cyclase System in Monocrotaline-treated Rats. J. Mol. Cell. Cardiol. 2000, 32, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Lu, Z.; Zhang, Y.; Geng, S.; Xu, M.; Xu, L.; Huang, Y.; Zhuang, P.; Zhang, Y. Stage-dependent changes of β2-adrenergic receptor signaling in right ventricular remodeling in monocrotaline-induced pulmonary arterial hypertension. Int. J. Mol. Med. 2018, 41, 2493–2504. [Google Scholar] [CrossRef]
- Vescovo, G.; Jones, S.M.; Harding, S.E.; Poole-Wilson, P.A. Isoproterenol sensitivity of isolated cardiac myocytes from rats with monocrotaline-induced right-sided hypertrophy and heart failure. J. Mol. Cell. Cardiol. 1989, 21, 1047–1061. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2015, 37, 67–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingwall, J.S.; Weiss, R.G. Is the Failing Heart Energy Starved? Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, P.A.; Panjrath, G.S.; Lai, S.; Hirsch, G.A.; Wu, K.; Najjar, S.S.; Steinberg, A.; Gerstenblith, G.; Weiss, R.G. Metabolic Rates of ATP Transfer Through Creatine Kinase (CK Flux) Predict Clinical Heart Failure Events and Death. Sci. Transl. Med. 2013, 5, 215re213. [Google Scholar] [CrossRef]
- Redout, E.M.; Wagner, M.J.; Zuidwijk, M.J.; Boer, C.; Musters, R.J.P.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 2007, 75, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Paulin, R.; Zervopoulos, S.; Haromy, A.; Nagendran, J.; Michelakis, E. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J. Mol. Med. 2013, 91, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Archer, S.L. The Right Ventricle in Pulmonary Arterial Hypertension: Disorders of Metabolism, Angiogenesis and Adrenergic Signaling in Right Ventricular Failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef]
- Potus, F.; Hindmarch, C.; Dunham-Snary, K.; Stafford, J.; Archer, S. Transcriptomic Signature of Right Ventricular Failure in Experimental Pulmonary Arterial Hypertension: Deep Sequencing Demonstrates Mitochondrial, Fibrotic, Inflammatory and Angiogenic Abnormalities. Int. J. Mol. Sci. 2018, 19, 2730. [Google Scholar] [CrossRef]
- Oikawa, M.; Kagaya, Y.; Otani, H.; Sakuma, M.; Demachi, J.; Suzuki, J.; Takahashi, T.; Nawata, J.; Ido, T.; Watanabe, J.; et al. Increased [18F]Fluorodeoxyglucose Accumulation in Right Ventricular Free Wall in Patients With Pulmonary Hypertension and the Effect of Epoprostenol. J. Am. Coll. Cardiol. 2005, 45, 1849–1855. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Fang, Y.-H.; Cadete, V.J.; Wietholt, C.; Urboniene, D.; Toth, P.; Marsboom, G.; Zhang, H.; Haber, I.; Rehman, J.; et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60. [Google Scholar] [CrossRef] [PubMed]
- van Wolferen, S.A.; Marcus, J.T.; Westerhof, N.; Spreeuwenberg, M.D.; Marques, K.M.J.; Bronzwaer, J.G.F.; Henkens, I.R.; Gan, C.T.-J.; Boonstra, A.; Postmus, P.E.; et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur. Heart J. 2008, 29, 120–127. [Google Scholar] [CrossRef]
- Anversa, P.; Ricci, R.; Olivetti, G. Effects of exercise on the capillary vasculature of the rat heart. Circulation 1987, 75, I12–I18. [Google Scholar] [PubMed]
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, E.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation 2015, 132, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.I.; Gomez-Arroyo, J.; Dumur, C.I.; Kraskauskas, D.; Natarajan, R.; Bogaard, H.J.; Fawcett, P.; Voelkel, N.F. Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol. Genom. 2013, 45, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Khuchua, Z.A.; Vassil’eva, E.V.; Medved’eva, N.V.; Saks, V.A. Heart mitochondrial creatine kinase revisited: The outer mitochondrial membrane is not important for coupling of phosphocreatine production to oxidative phosphorylation. Arch. Biochem. Biophys. 1989, 268, 176–190. [Google Scholar] [CrossRef]
- Bessman, S.P.; Fonyo, A. The possible role of the mitochondrial bound creatine kinase in regulation of mitochondrial respiration. Biochem. Biophys. Res. Commun. 1966, 22, 597–602. [Google Scholar] [CrossRef]
- Takahashi, E. Anoxic cell core can promote necrotic cell death in cardiomyocytes at physiological extracellular Po2. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H2507–H2515. [Google Scholar] [CrossRef]
- Ishikawa, K.; Hashimoto, H.; Mitani, S.; Toki, Y.; Okumura, K.; Ito, T. Enalapril improves heart failure induced by monocrotaline without reducing pulmonary hypertension in rats: Roles of preserved myocardial creatine kinase and lactate dehydrogenase isoenzymes. Int. J. Cardiol. 1995, 47, 225–233. [Google Scholar] [CrossRef]
- Fowler, E.D.; Benoist, D.; Drinkhill, M.J.; Stones, R.; Helmes, M.; Wüst, R.C.I.; Stienen, G.J.M.; Steele, D.S.; White, E. Decreased creatine kinase is linked to diastolic dysfunction in rats with right heart failure induced by pulmonary artery hypertension. J. Mol. Cell. Cardiol. 2015, 86, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Joubert, F.; Wilding, J.R.; Fortin, D.; Domergue-Dupont, V.; Novotova, M.; Ventura-Clapier, R.; Veksler, V. Local energetic regulation of sarcoplasmic and myosin ATPase is differently impaired in rats with heart failure. J. Physiol. 2008, 586, 5181–5192. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.; Veksler, V.; Minajeva, A.; Kaasik, A.; Mateo, P.; Mayoux, E.; Hoerter, J.; Bigard, X.; Serrurier, B.; Ventura-Clapier, R. Subcellular Creatine Kinase Alterations. Circ. Res. 1999, 85, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, P.P.-S.; Davies, R.A.; Chandy, G.; Stewart, D.; Beanlands, R.S.B.; Haddad, H.; Pugliese, C.; Mielniczuk, L.M. Usefulness of Beta-Blocker Therapy and Outcomes in Patients with Pulmonary Arterial Hypertension. Am. J. Cardiol. 2012, 109, 1504–1509. [Google Scholar] [CrossRef]
- Thenappan, T.; Roy, S.S.; Duval, S.; Glassner-Kolmin, C.; Gomberg-Maitland, M. β-Blocker Therapy Is Not Associated with Adverse Outcomes in Patients With Pulmonary Arterial Hypertension: A Propensity Score Analysis. Circ. Heart Fail. 2014, 7, 903–910. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Bajaj, N.S.; Zein, J.; Minai, O.A.; Dweik, R.A. Outcomes of β-blocker use in pulmonary arterial hypertension: A propensity-matched analysis. Eur. Respir. J. 2015, 46, 750–760. [Google Scholar] [CrossRef]
- Farha, S.; Saygin, D.; Park, M.M.; Cheong, H.I.; Asosingh, K.; Comhair, S.A.A.; Stephens, O.R.; Roach, E.C.; Sharp, J.; Highland, K.B.; et al. Pulmonary arterial hypertension treatment with carvedilol for heart failure: A randomized controlled trial. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Natarajan, R.; Mizuno, S.; Abbate, A.; Chang, P.J.; Chau, V.Q.; Hoke, N.N.; Kraskauskas, D.; Kasper, M.; Salloum, F.N.; et al. Adrenergic Receptor Blockade Reverses Right Heart Remodeling and Dysfunction in Pulmonary Hypertensive Rats. Am. J. Respir. Critical Care Med. 2010, 182, 652–660. [Google Scholar] [CrossRef]
- de Man, F.S.; Handoko, M.L.; van Ballegoij, J.J.M.; Schalij, I.; Bogaards, S.J.P.; Postmus, P.E.; van der Velden, J.; Westerhof, N.; Paulus, W.J.; Vonk-Noordegraaf, A. Bisoprolol Delays Progression Towards Right Heart Failure in Experimental Pulmonary Hypertension. Circ. Heart Fail. 2012, 5, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Perros, F.; Ranchoux, B.; Izikki, M.; Bentebbal, S.; Happé, C.; Antigny, F.; Jourdon, P.; Dorfmüller, P.; Lecerf, F.; Fadel, E.; et al. Nebivolol for Improving Endothelial Dysfunction, Pulmonary Vascular Remodeling, and Right Heart Function in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2015, 65, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Fowler, E.D.; Drinkhill, M.J.; Norman, R.; Pervolaraki, E.; Stones, R.; Steer, E.; Benoist, D.; Steele, D.S.; Calaghan, S.C.; White, E. Beta1-adrenoceptor antagonist, metoprolol attenuates cardiac myocyte Ca2+ handling dysfunction in rats with pulmonary artery hypertension. J. Mol. Cell. Cardiol. 2018, 120, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Fowler, E.D.; Drinkhill, M.J.; Stones, R.; White, E. Diastolic dysfunction in pulmonary artery hypertension: Creatine kinase and the potential therapeutic benefit of beta-blockers. Clin. Exp. Pharmacol. Physiol. 2018, 45, 384–389. [Google Scholar] [CrossRef]
- Al-Shammari, A.A.; Gaffney, E.A.; Egginton, S. Re-evaluating the Use of Voronoi Tessellations in the Assessment of Oxygen Supply from Capillaries in Muscle. Bull. Math. Biol. 2012, 74, 2204–2231. [Google Scholar] [CrossRef] [Green Version]
- Al-Shammari, A.A.; Gaffney, E.A.; Egginton, S. Modelling capillary oxygen supply capacity in mixed muscles: Capillary domains revisited. J. Theor. Biol. 2014, 356, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Al-Shammari, A.A.; Kissane, R.W.P.; Holbek, S.; Mackey, A.L.; Andersen, T.R.; Gaffney, E.A.; Kjaer, M.; Egginton, S. An integrated method for quantitative morphometry and oxygen transport modelling in striated muscle. J. Appl. Physiol. 2018, 126, 544–557. [Google Scholar] [CrossRef]
- Goldman, D. Theoretical Models of Microvascular Oxygen Transport to Tissue. Microcirculation 2008, 15, 795–811. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, H.; Sharov, V.; Lesch, M.; Goldstein, S. Progression of heart failure: A role for interstitial fibrosis. Mol. Cell. Biochem. 1995, 147, 29–34. [Google Scholar] [CrossRef]
- van der Laarse, W.J.; des Tombe, A.L.; van Beek-Harmsen, B.J.; Lee-de Groot, M.B.; Jaspers, R.T. Krogh’s diffusion coefficient for oxygen in isolated Xenopus skeletal muscle fibers and rat myocardial trabeculae at maximum rates of oxygen consumption. J. Appl. Physiol. 2005, 99, 2173–2180. [Google Scholar] [CrossRef]
- Hauton, D.; Al-Shammari, A.; Gaffney, E.A.; Egginton, S. Maternal Hypoxia Decreases Capillary Supply and Increases Metabolic Inefficiency Leading to Divergence in Myocardial Oxygen Supply and Demand. PLoS ONE 2015, 10, e0127424. [Google Scholar] [CrossRef]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [Green Version]
- Kaasik, A.; Veksler, V.; Boehm, E.; Novotova, M.; Minajeva, A.; Ventura-Clapier, R. Energetic Crosstalk Between Organelles: Architectural Integration of Energy Production and Utilization. Circ. Res. 2001, 89, 153–159. [Google Scholar] [CrossRef]
- Partovian, C.; Adnot, S.; Eddahibi, S.; Teiger, E.; Levame, M.; Dreyfus, P.; Raffestin, B.; Frelin, C. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am. J. Physiol. 1998, 275, H1948–H1956. [Google Scholar] [CrossRef]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar]
- Anand, V.; Roy, S.S.; Archer, S.L.; Weir, E.K.; Garg, S.K.; Duval, S.; Thenappan, T. Trends and Outcomes of Pulmonary Arterial Hypertension-Related Hospitalizations in the United States: Analysis of the Nationwide Inpatient Sample Database From 2001 Through 2012. JAMA Cardiol. 2016, 1, 1021–1029. [Google Scholar] [CrossRef]
- Shimony, A.; Eisenberg, M.J.; Rudski, L.G.; Schlesinger, R.; Afilalo, J.; Joyal, D.; Dragatakis, L.; Hirsch, A.; Boutet, K.; Fox, B.D.; et al. Prevalence and impact of coronary artery disease in patients with pulmonary arterial hypertension. Am. J. Cardiol. 2011, 108, 460–464. [Google Scholar] [CrossRef]
- Enbergs, A.; Burger, R.; Reinecke, H.; Borggrefe, M.; Breithardt, G.; Kerber, S. Prevalence of coronary artery disease in a general population without suspicion of coronary artery disease: Angiographic analysis of subjects aged 40 to 70 years referred for catheter ablation therapy. Eur. Heart J. 2000, 21, 45–52. [Google Scholar] [CrossRef]
- Meloche, J.; Lampron, M.C.; Nadeau, V.; Maltais, M.; Potus, F.; Lambert, C.; Tremblay, E.; Vitry, G.; Breuils-Bonnet, S.; Boucherat, O.; et al. Implication of Inflammation and Epigenetic Readers in Coronary Artery Remodeling in Patients with Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1513–1523. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; McDonald, K.M.; Hastie, T.; Fadel, B.; Hagan, V.; Lee, B.K.; Hlatky, M.A. Meta-analysis of trials comparing beta-blockers, calcium antagonists, and nitrates for stable angina. JAMA 1999, 281, 1927–1936. [Google Scholar] [CrossRef]
- Alleman, R.J.; Stewart, L.M.; Tsang, A.M.; Brown, D.A. Why Does Exercise “Trigger” Adaptive Protective Responses in the Heart? Dose-Response 2015, 13. [Google Scholar] [CrossRef]
- Egginton, S. Invited review: Activity-induced angiogenesis. Pflüg. Arch.-Eur. J. Physiol. 2008, 457, 963. [Google Scholar] [CrossRef]
- Natali, A.J.; Fowler, E.D.; Calaghan, S.; White, E. Voluntary exercise delays heart failure onset in rats with pulmonary artery hypertension. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 421–424. [Google Scholar] [CrossRef]
- de Graaf, R.A.; van Kranenburg, A.; Nicolay, K. In vivo 31P-NMR diffusion spectroscopy of ATP and phosphocreatine in rat skeletal muscle. Biophys. J. 2000, 78, 1657–1664. [Google Scholar] [CrossRef]
- Birkedal, R.; Laasmaa, M.; Vendelin, M. The location of energetic compartments affects energetic communication in cardiomyocytes. Front. Physiol. 2014, 5, 376. [Google Scholar] [CrossRef]
- Wüst, R.C.I.; de Vries, H.J.; Wintjes, L.T.; Rodenburg, R.J.; Niessen, H.W.M.; Stienen, G.J.M. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc. Res. 2016, 111, 362. [Google Scholar] [CrossRef]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; et al. Myocardial Phosphocreatine-to-ATP Ratio Is a Predictor of Mortality in Patients with Dilated Cardiomyopathy. Circulation 1997, 96, 2190–2196. [Google Scholar] [CrossRef]
- Lamberts, R.R.; Caldenhoven, E.; Lansink, M.; Witte, G.; Vaessen, R.J.; St Cyr, J.A.; Stienen, G.J.M. Preservation of diastolic function in monocrotaline-induced right ventricular hypertrophy in rats. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H1869–H1876. [Google Scholar] [CrossRef]
- Lygate, C.A.; Bohl, S.; ten Hove, M.; Faller, K.M.E.; Ostrowski, P.J.; Zervou, S.; Medway, D.J.; Aksentijevic, D.; Sebag-Montefiore, L.; Wallis, J.; et al. Moderate elevation of intracellular creatine by targeting the creatine transporter protects mice from acute myocardial infarction. Cardiovasc. Res. 2012, 96, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.Y.; Handoko, M.L.; Mouchaers, K.T.B.; de Man, F.S.; Vonk-Noordegraaf, A.; van der Laarse, W.J. Reduced mechanical efficiency of rat papillary muscle related to degree of hypertrophy of cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1190–H1197. [Google Scholar] [CrossRef]
- Pham, T.; Nisbet, L.; Taberner, A.; Loiselle, D.; Han, J.C. Pulmonary arterial hypertension reduces energy efficiency of right, but not left, rat ventricular trabeculae. J. Physiol. 2018, 596, 1153–1166. [Google Scholar] [CrossRef]
- Roth, R.A.; Dotzlaf, L.A.; Baranyi, B.; Kuo, C.H.; Hook, J.B. Effect of monocrotaline ingestion on liver, kidney, and lung of rats. Toxicol. Appl. Pharm. 1981, 60, 193–203. [Google Scholar] [CrossRef]
- Sugita, T.; Hyers, T.M.; Dauber, I.M.; Wagner, W.W.; McMurtry, I.F.; Reeves, J.T. Lung vessel leak precedes right ventricular hypertrophy in monocrotaline-treated rats. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 54, 371–374. [Google Scholar] [CrossRef]
- Hardziyenka, M.; Campian, M.E.; Rianne de Bruin-Bon, H.A.C.M.; Michel, M.C.; Tan, H.L. Sequence of Echocardiographic Changes During Development of Right Ventricular Failure in Rat. J. Am. Soc. Echocardiogr. 2006, 19, 1272–1279. [Google Scholar] [CrossRef]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The Monocrotaline Model of Pulmonary Hypertension in Perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 302, L363–L369. [Google Scholar] [CrossRef]
- Maarman, G.; Lecour, S.; Butrous, G.; Thienemann, F.; Sliwa, K. A comprehensive review: The evolution of animal models in pulmonary hypertension research; are we there yet? Pulm. Circ. 2013, 3, 739–756. [Google Scholar] [CrossRef]
- Ryan, J.J.; Marsboom, G.; Archer, S.L. Rodent Models of Group 1 Pulmonary Hypertension. In Pharmacotherapy of Pulmonary Hypertension; Humbert, M., Evgenov, O.V., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 105–149. [Google Scholar] [CrossRef]
- Nogueira-Ferreira, R.; Vitorino, R.; Ferreira, R.; Henriques-Coelho, T. Exploring the monocrotaline animal model for the study of pulmonary arterial hypertension: A network approach. Pulm. Pharmacol. Ther. 2015, 35, 8–16. [Google Scholar] [CrossRef]
- Beard, D.A.; Schenkman, K.A.; Feigl, E.O. Myocardial oxygenation in isolated hearts predicted by an anatomically realistic microvascular transport model. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1826–H1836. [Google Scholar] [CrossRef]
- McCrossan, Z.A.; Billeter, R.; White, E. Transmural changes in size, contractile and electrical properties of SHR left ventricular myocytes during compensated hypertrophy. Cardiovasc. Res. 2004, 63, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.E.; Jarvis, K.L.; Hyland, K.J. Protein measurement using bicinchoninic acid: Elimination of interfering substances. Anal. Biochem. 1989, 180, 136–139. [Google Scholar] [CrossRef]
- Rosalki, S.B. An improved procedure for serum creatine phosphokinase determination. J. Lab. Clin. Med. 1967, 69, 696–705. [Google Scholar]
- Srere, P.A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. In Methods in Enzymology; John, M.L., Ed.; Academic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 3–11. [Google Scholar]





{kind=link}
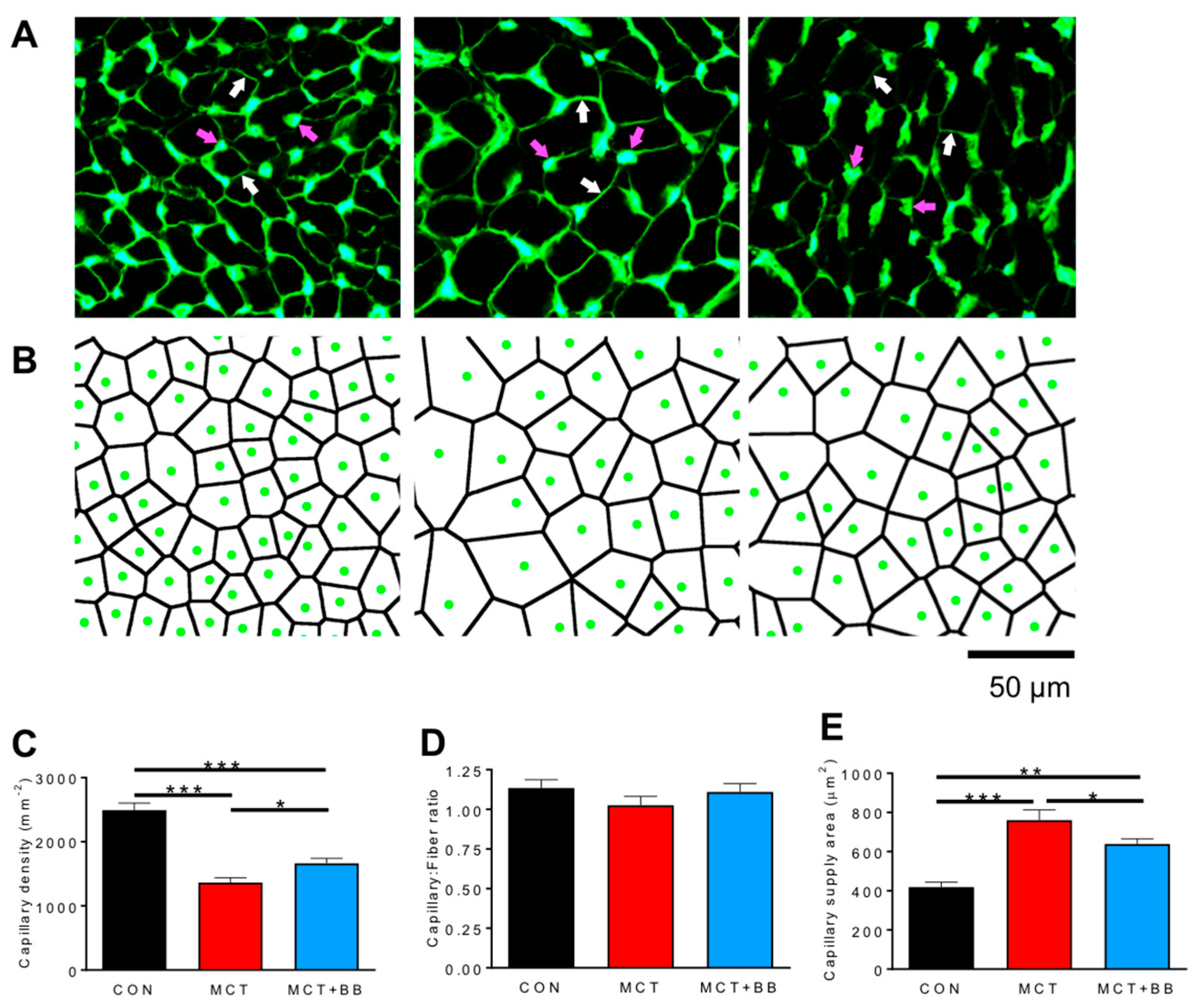{kind=link}
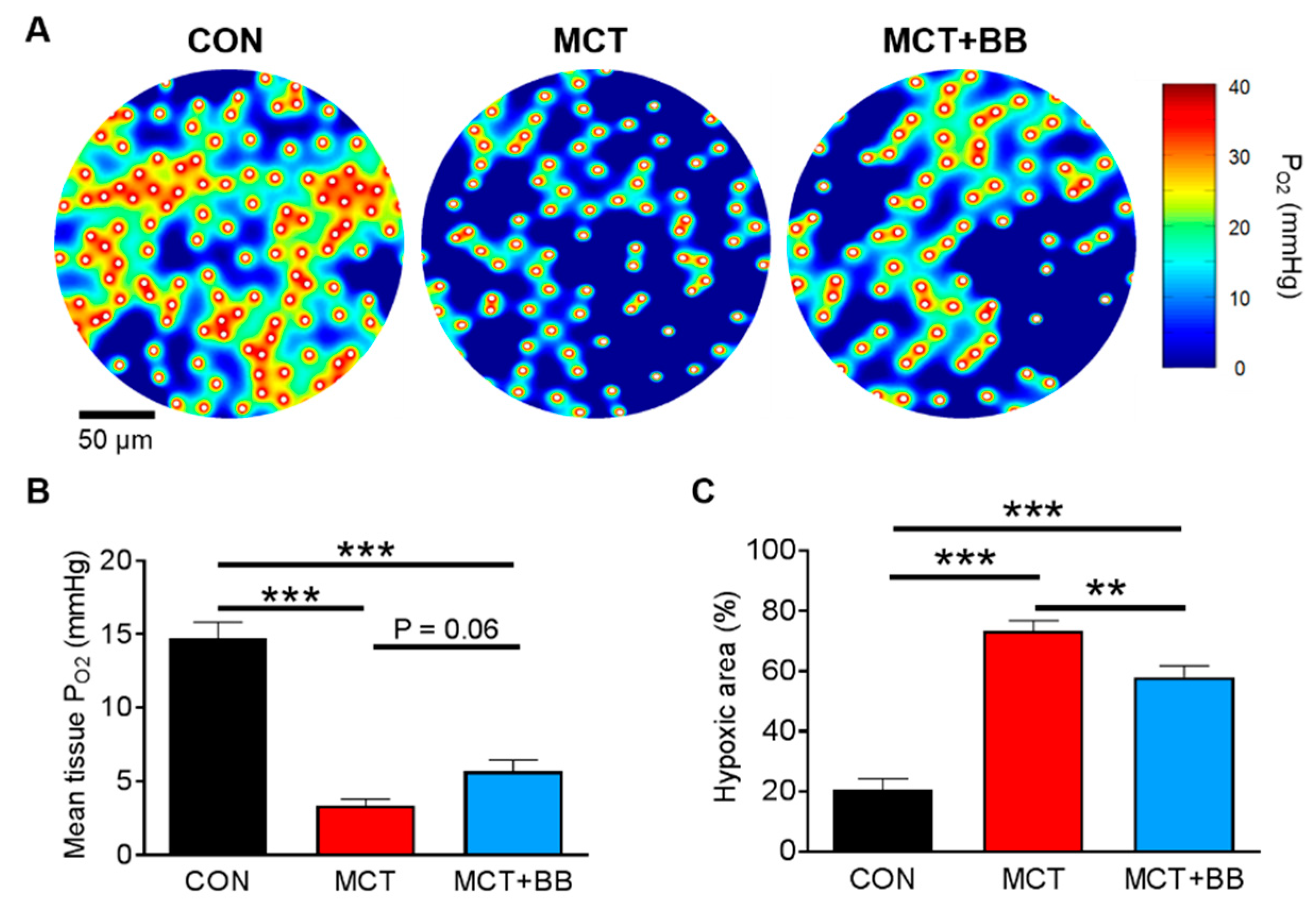{kind=link}
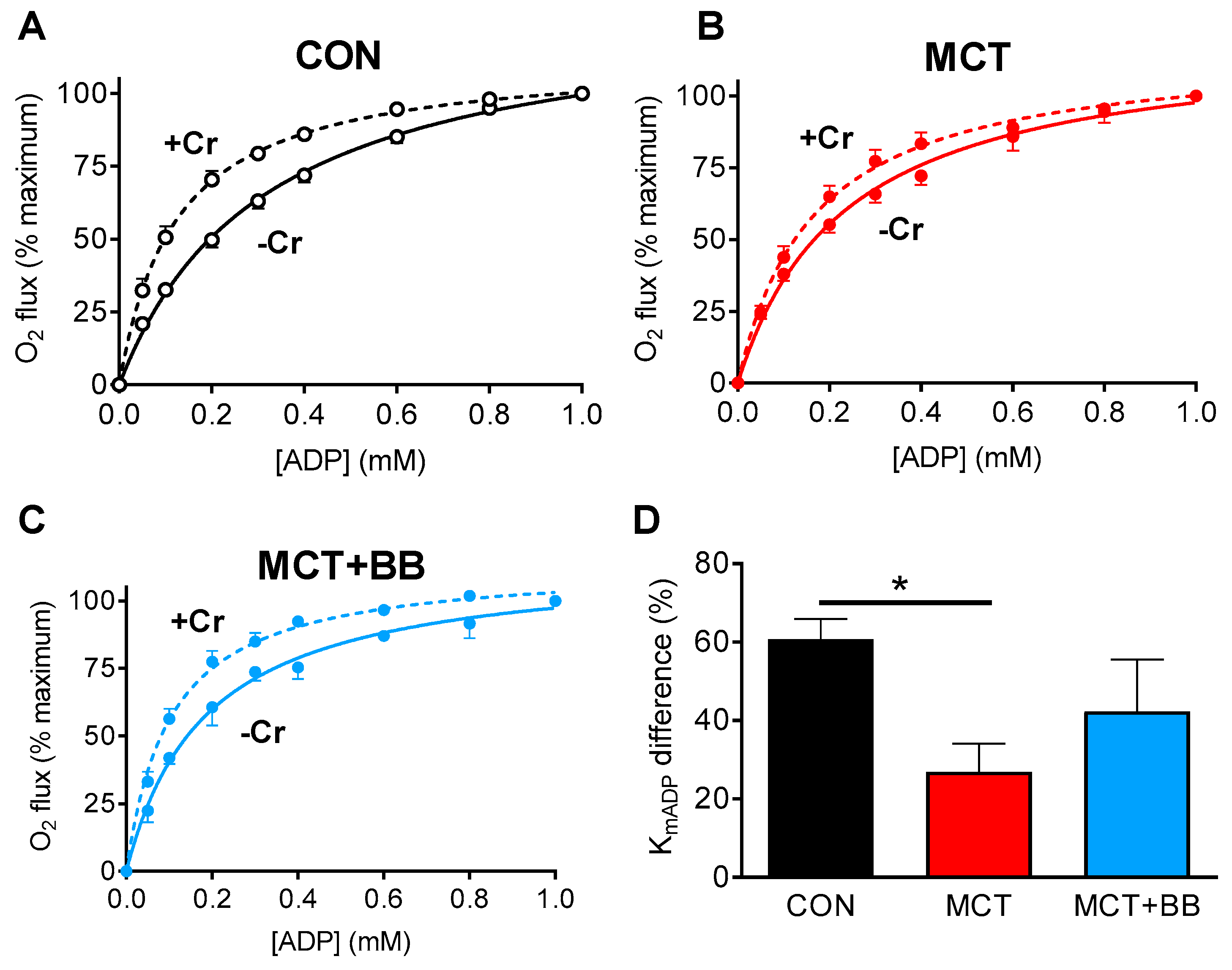{kind=link}
{kind=link}
| CON | MCT | MCT+BB | |
|---|---|---|---|
| Body weight (g) | 330 ± 7 | 294 ± 13 | 273 ± 10 ** |
| HW:BW (mg/g) | 3.90 ± 0.13 | 6.22 ± 0.34 *** | 5.07 ± 0.34 *,† |
| RV:BW (mg/g) | 0.75 ± 0.05 | 1.42 ± 0.12 *** | 0.90 ± 0.07 †† |
| LV + S:BW (mg/g) | 2.78 ± 0.17 | 3.00 ± 0.19 | 2.96 ± 0.09 |
| Lung:BW (mg/g) | 7.53 ± 0.82 | 10.3 ± 0.3 * | 10.2 ± 0.6 * |
| Liver:BW (mg/g) | 39.6 ± 1.3 | 40.6 ± 2.8 | 49.2 ± 1.8 *,† |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fowler, E.D.; Hauton, D.; Boyle, J.; Egginton, S.; Steele, D.S.; White, E. Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System. Int. J. Mol. Sci. 2019, 20, 1805. https://doi.org/10.3390/ijms20081805
Fowler ED, Hauton D, Boyle J, Egginton S, Steele DS, White E. Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System. International Journal of Molecular Sciences. 2019; 20(8):1805. https://doi.org/10.3390/ijms20081805
Chicago/Turabian StyleFowler, Ewan D., David Hauton, John Boyle, Stuart Egginton, Derek S. Steele, and Ed White. 2019. "Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System" International Journal of Molecular Sciences 20, no. 8: 1805. https://doi.org/10.3390/ijms20081805
APA StyleFowler, E. D., Hauton, D., Boyle, J., Egginton, S., Steele, D. S., & White, E. (2019). Energy Metabolism in the Failing Right Ventricle: Limitations of Oxygen Delivery and the Creatine Kinase System. International Journal of Molecular Sciences, 20(8), 1805. https://doi.org/10.3390/ijms20081805