CD20-Mimotope Peptides: A Model to Define the Molecular Basis of Epitope Spreading

Abstract
:1. Introduction
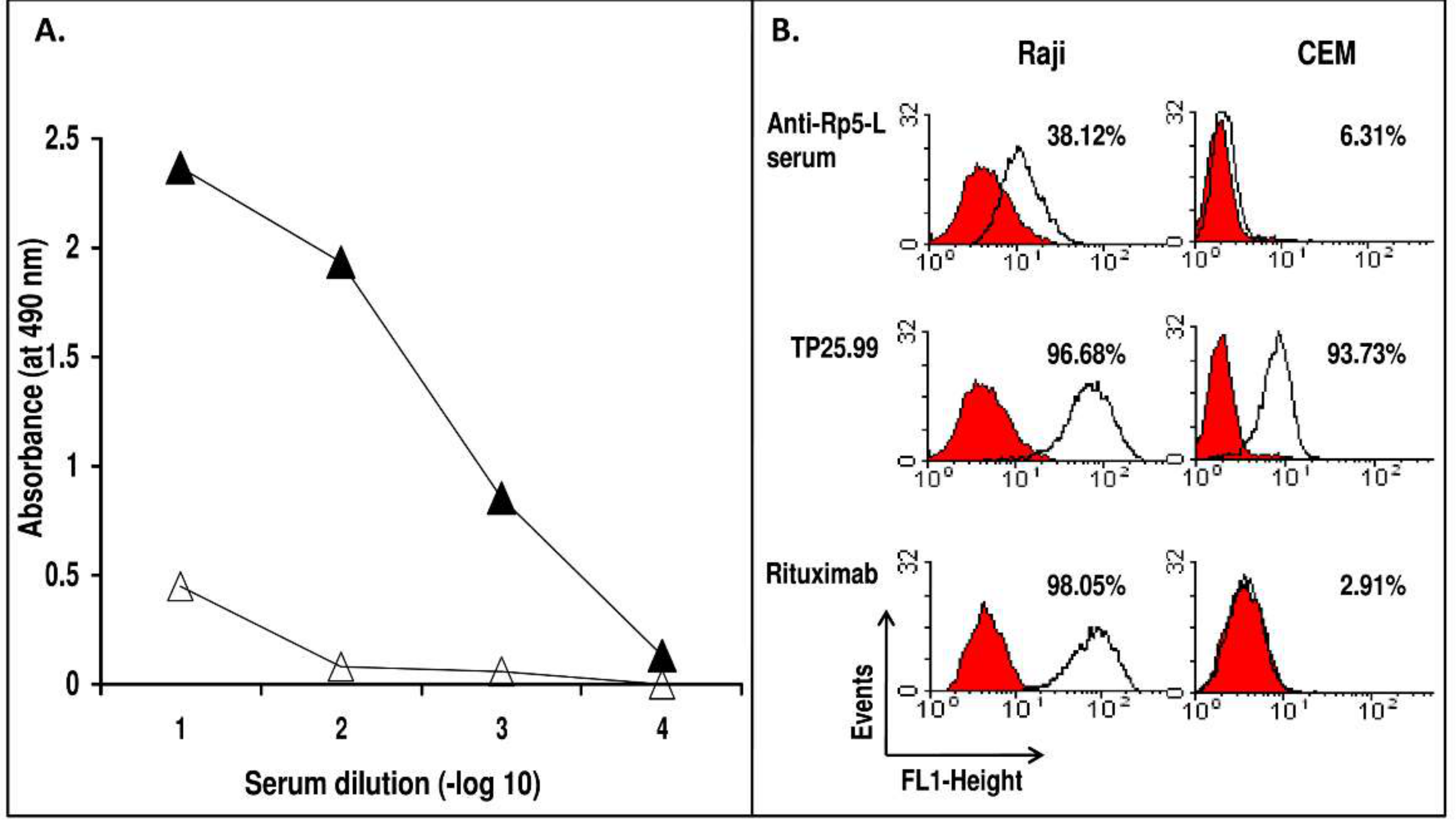
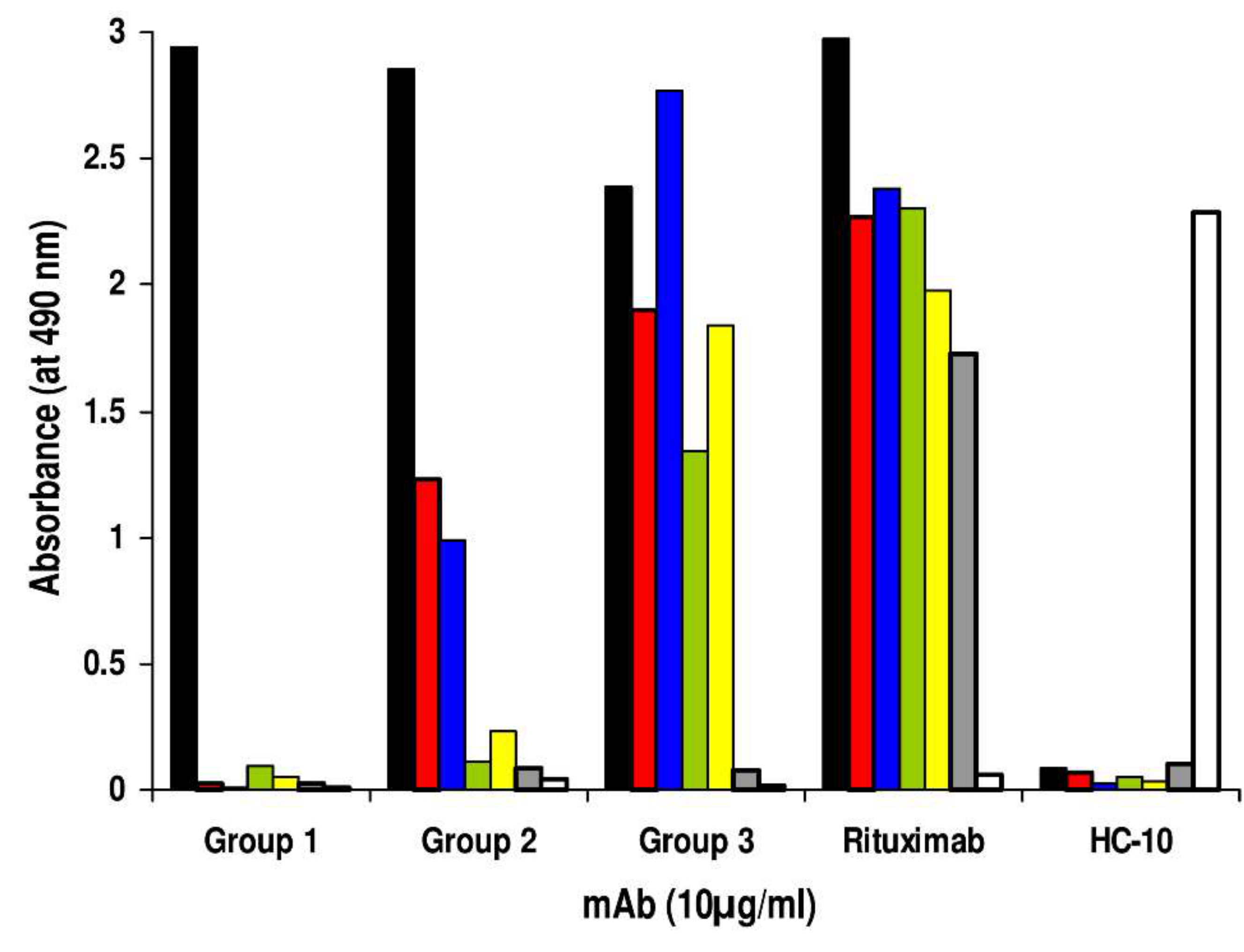
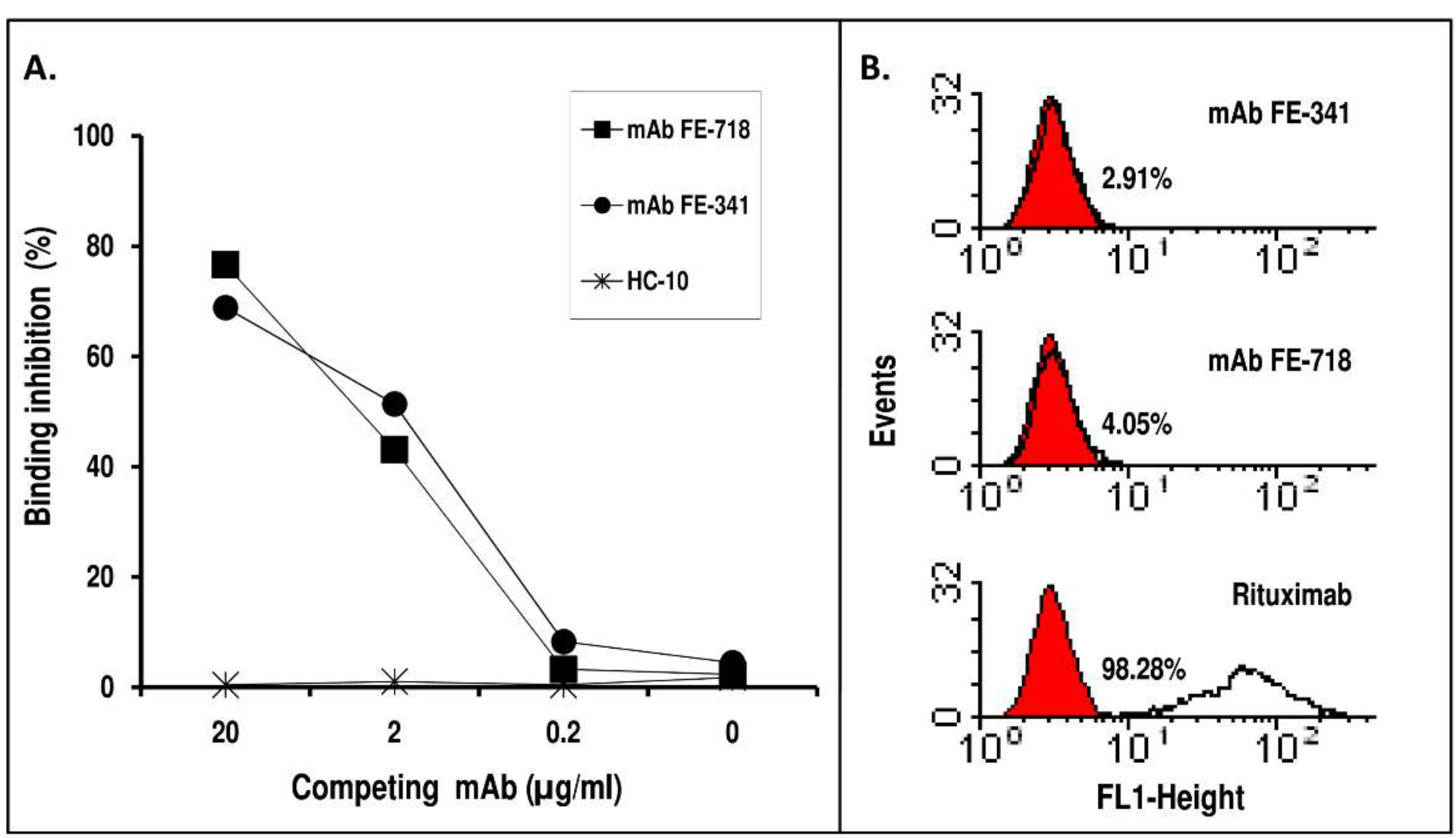
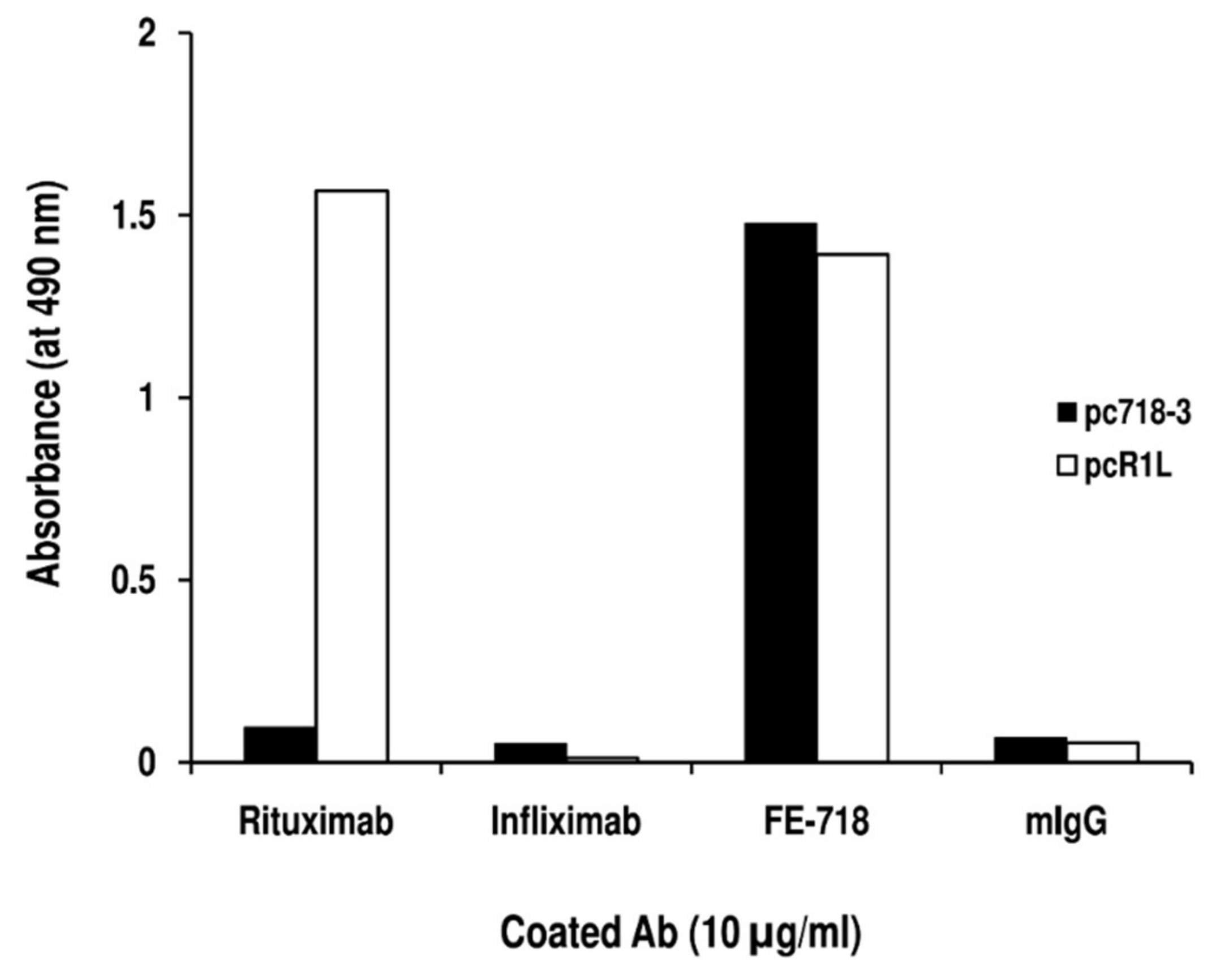
2. Results
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cells
4.3. Conventional Reagents, mAb, Peptides and PDPL
4.4. Preparation of Anti-Rp5-L mAb
4.5. Binding Assay
4.6. Flow Cytometry
4.7. Biopanning, Immunoscreening, and Sequence Analysis
Author Contributions
Funding
Acknowledgments
Conflict of Interest
References
- Bucktrout, S.L.; Bluestone, J.A.; Ramsdell, F. Recent advances in immunotherapies: From infection and autoimmunity, to cancer, and back again. Genome Med. 2018, 10, 79. [Google Scholar] [CrossRef]
- Alfonso, M.; Díaz, A.; Hernández, A.M.; Pérez, A.; Rodríguez, E.; Bitton, R.; Pérez, R.; Vázquez, A.M. An anti-idiotype vaccine elicits a specific response to N-glycolyl sialic acid residues of glycoconjugates in melanoma patients. J. Immunol. 2002, 168, 2523–2529. [Google Scholar] [CrossRef]
- De Cerio, A.L.; Zabalegui, N.; Rodriguez-Calvillo, M.; Inoges, S.; Bendandi, M. Anti-idiotype antibodies in cancer treatment. Oncogene 2007, 26, 3594–3602. [Google Scholar] [CrossRef] [Green Version]
- Kieber-Emmons, T.; Monzavi-Karbassi, B.; Pashov, A.; Saha, S.; Murali, R.; Kohler, H. The promise of the anti-idiotype concept. Front. Oncol. 2012, 2, 196. [Google Scholar] [CrossRef]
- Ladjemi, M.Z. Anti-idiotypic antibodies as cancer vaccines: Achievements and future improvements. Front. Oncol. 2012, 2, 158. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.M.; Rodreguez-Zhurbenko, N.; Lopez, A.M. Anti-ganglioside anti-idiotypic vaccination: More than molecular mimicry. Front. Oncol. 2012, 2, 170. [Google Scholar] [CrossRef] [PubMed]
- Cacciavillano, W.; Sampor, C.; Venier, C.; Gabri, M.R.; de Dávila, M.T.; Galluzzo, M.L.; Guthmann, M.D.; Fainboim, L.; Alonso, D.F.; Chantada, G.L. A Phase I Study of the Anti-Idiotype Vaccine Racotumomab in Neuroblastoma and Other Pediatric Refractory Malignancies. Pediatr. Blood Cancer 2015, 62, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Yada, E.; Ohtake, J.; Fujimoto, Y.; Uchiyama, H.; Yoshida, S.; Sasada, T. Current status and future prospects of peptide-based cancer vaccines. Immunotherapy 2016, 8, 1321–1333. [Google Scholar] [CrossRef]
- Klausen, U.; Holmberg, S.; Holmström, M.O.; Jørgensen, N.G.D.; Grauslund, J.H.; Svane, I.M.; Andersen, M.H. Novel Strategies for Peptide-Based Vaccines in Hematological Malignancies. Front. Immunol. 2018, 9, 2264. [Google Scholar] [CrossRef] [PubMed]
- Obara, W.; Kanehira, M.; Katagiri, T.; Kato, R.; Kato, Y.; Takata, R. Present status and future perspective of peptide-based vaccine therapy for urological cancer. Cancer Sci. 2018, 109, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Shi, J.; Yang, C. HER2-Based Immunotherapy for Breast Cancer. Cancer Biother. Radiopharm. 2018, 33, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Prete, M.; Perosa, F.; Favoino, E.; Dammacco, F. Biological therapy with monoclonal antibodies: A novel treatment approach to autoimmune disease. Clin. Exp. Med. 2005, 5, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Prete, M.; Racanelli, V.; Dammacco, F. CD20-depleting therapy in autoimmune diseases: From basic research to the clinic. J. Intern. Med. 2010, 267, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.K.; Livingston, P.O.; Agus, D.B.; Pinilla-Ibarz, J.; Zelenetz, A.; Scheinberg, D.A. Vaccination with CD20 peptides induces a biologically active, specific immune response in mice. Blood 2002, 99, 3748–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Prete, M.; Dammacco, F. CD20: A target antigen for immunotherapy of autoimmune diseases. Autoimmun. Rev. 2005, 4, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Dammacco, F. CD20 mimicry by a mAb rituximab-specific linear peptide: A potential tool for active immunotherapy of autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1051, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Dammacco, F. Generation of biologically active linear and cyclic peptides has revealed a unique fine specificity of rituximab and its possible cross-reactivity with acid sphingomyelinase-like phosphodiesterase 3b precursor. Blood 2006, 107, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Favoino, E.; Vicenti, C.; Guarnera, A.; Racanelli, V.; De Pinto, V.; Dammacco, F. Two structurally different rituximab-specific CD20 mimotope peptides reveal that rituximab recognizes two different CD20-associated epitopes. J. Immunol. 2009, 182, 416–423. [Google Scholar] [CrossRef]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef]
- Perosa, F.; Favoino, E.; Vicenti, C.; Merchionne, F.; Dammacco, F. Identification of an antigenic and immunogenic motif expressed by two 7-mer rituximab-specific cyclic peptide mimotopes: Implication for peptide-based active immunotherapy. J. Immunol. 2007, 179, 7967–7974. [Google Scholar] [CrossRef]
- Favoino, E.; Prete, M.; Marzullo, A.; Millo, E.; Shoenfeld, Y.; Perosa, F. CD20-Mimotope Peptide Active Immunotherapy in Systemic Lupus Erythematosus and a Reappraisal of Vaccination Strategies in Rheumatic Diseases. Clin. Rev. Allergy Immunol. 2017, 52, 217–233. [Google Scholar] [CrossRef]
- Hou, Y.; Gu, X.X. Development of peptide mimotopes of lipooligosaccharide from nontypeable Haemophilus influenzae as vaccine candidates. J. Immunol. 2003, 170, 4373–4379. [Google Scholar] [CrossRef]
- Wagner, S.; Hafner, C.; Allwardt, D.; Jasinska, J.; Ferrone, S.; Zielinski, C.C.; Scheiner, O.; Wiedermann, U.; Pehamberger, H.; Breiteneder, H. Vaccination with a human high molecular weight melanoma-associated antigen mimotope induces a humoral response inhibiting melanoma cell growth in vitro. J. Immunol. 2005, 174, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Krepler, C.; Allwardt, D.; Latzka, J.; Strommer, S.; Scheiner, O.; Pehamberger, H.; Wiedermann, U.; Hafner, C.; Breiteneder, H. Reduction of human melanoma tumor growth in severe combined immunodeficient mice by passive transfer of antibodies induced by a high molecular weight melanoma-associated antigen mimotope vaccine. Clin. Cancer Res. 2008, 14, 8178–8183. [Google Scholar] [CrossRef] [PubMed]
- Vanderlugt, C.L.; Miller, S.D. Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Goodell, V.; Schiffman, K.; Knutson, K.L. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J. Clin. Immunol. 2004, 24, 571–578. [Google Scholar] [CrossRef]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.-Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulley, J.L.; Madan, R.A.; Pachynski, R.; Mulders, P.; Sheikh, N.A.; Trager, J.; Drake, C.G. Role of Antigen Spread and Distinctive Characteristics of Immunotherapy in Cancer Treatment. J. Natl. Cancer Inst. 2017, 109, djw261. [Google Scholar] [CrossRef]
- Vanderlugt, C.J.; Miller, S.D. Epitope spreading. Curr. Opin. Immunol. 1996, 8, 831–836. [Google Scholar] [CrossRef]
- Latzka, J.; Gaier, S.; Hofstetter, G.; Balazs, N.; Smole, U.; Ferrone, S.; Scheiner, O.; Breiteneder, H.; Pehamberger, H.; Wagner, S. Specificity of mimotope-induced anti-high molecular weight-melanoma associated antigen (HMW-MAA) antibodies does not ensure biological activity. PLoS ONE 2011, 6, e19383. [Google Scholar] [CrossRef]
- Beenhouwer, D.O.; May, R.J.; Valadon, P.; Scharff, M.D. High affinity mimotope of the polysaccharide capsule of Cryptococcus neoformans identified from an evolutionary phage peptide library. J. Immunol. 2002, 169, 6992–6999. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hsu, J.C.; Kieber-Emmons, T.; Wang, X.; Ferrone, S. Human tumor associated antigen mimicry by xenoantigens, anti-idiotypic antibodies and peptide mimics: Implications for immunotherapy of malignant diseases. Cancer Chemother. Biol. Response Modif. 2005, 22, 769–787. [Google Scholar] [PubMed]
- Van Regenmortel, M.H. Molecular design versus empirical discovery in peptide-based vaccines. Coming to terms with fuzzy recognition sites and ill-defined structure-function relationships in immunology. Vaccine 1999, 18, 216–221. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H. Antigenicity and immunogenicity of synthetic peptides. Biologicals 2001, 29, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.J.; Katsumata, Y.; Ascherman, D.P. Structural and thermodynamic approach to peptide immunogenicity. PLoS Comput. Biol. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Schiffman, K.; Guthrie, K.; Salazar, L.G.; Knutson, K.L.; Goodell, V.; dela Rosa, C.; Cheever, M.A. Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein—Based vaccine. J. Clin. Oncol. 2004, 22, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Polyak, M.J.; Deans, J.P. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood 2002, 99, 3256–3262. [Google Scholar] [CrossRef] [Green Version]
- Niederfellner, G.; Lammens, A.; Mundigl, O.; Georges, G.J.; Schaefer, W.; Schwaiger, M.; Franke, A.; Wiechmann, K.; Jenewein, S.; Slootstra, J.W.; et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011, 118, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhao, Y.; He, W.; Wang, W.; Chen, Y.; Zhang, S.; Ma, Y.; Gohda, J.; Ishida, T.; Walter, T.S.; et al. A RANKL mutant used as an inter-species vaccine for efficient immunotherapy of osteoporosis. Sci. Rep. 2015, 5, 14150. [Google Scholar] [CrossRef]
- Perosa, F.; Luccarelli, G.; Prete, M.; Favoino, E.; Ferrone, S.; Dammacco, F. Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J. Immunol. 2003, 171, 1918–1926. [Google Scholar] [CrossRef]
- Perosa, F.; Carbone, R.; Ferrone, S.; Dammacco, F. Purification of human immunoglobulins by sequential precipitation with caprylic acid and ammonium sulphate. J. Immunol. Methods 1990, 128, 9–16. [Google Scholar] [CrossRef]
- Favoino, E.; Digiglio, L.; Cuomo, G.; Favia, I.E.; Racanelli, V.; Valentini, G.; Perosa, F. Autoantibodies recognizing the amino terminal 1–17 segment of CENP-A display unique specificities in systemic sclerosis. PLoS ONE 2013, 8, e61453. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Origin | Peptide | |
|---|---|---|
| Denomination | Sequence | |
| Rituximab-specific peptides | ||
| Rp5-L | QDKLTQWPKWLEg | |
| Rp1-L | WPRWLEN | |
| Rp10-L | ITPWPHWLERSSg | |
| ASMLPD | ||
| Rev-pASMLPD | 163SLWPKWLEAIQ153 | |
| pASMLPD | 153QIAELWKPWLS163 | |
| Human CD20 | ||
| RpCD20-L | 165YNCEPANPSEKNSPSTQYCY184 |
| Phage Clone Insert # | Clones, n (%) | Deduced Amino Acid Insert Sequence (a) | Specificity of Reactivity (A490nm) | |
|---|---|---|---|---|
| mAb | mIgG | |||
| pc718-1 | 5 (27.7%) | WPHVLPE | 1.754 ± 0.01 | 0.123 ± 0.002 |
| pc718-2 | 2 (11.1%) | KWPQYLS | 1.833 ± 0.14 | 0.144 ± 0.07 |
| pc718-3 | 11 (61.1%) | MWPKWLP | 1.92 ± 0.044 | 0.107 ± 0.02 |
| FE-718 motif | WP—-L | |||
| pc341-1 | 4 (22.2%) | SLKMPHWPHLLP | 1.644 ± 0.01 | 0.167 ± 0.002 |
| pc341-2 | 1 (5.5%) | QHVNLARWPWQL | 1.834 ± 0.021 | 0.111 ± 0.013 |
| pc341-3 | 10 (55.5%) | TQLGWPHSIGDA | 1.421 ± 0.08 | 0.172 ± 0.1 |
| pc341-4 | 2 (11.1%) | HSSWPRHLDPPQ | 1.962 ± 0.013 | 0.069 ± 0.002 |
| pc341-5 | 1 (5.5%) | QWPNELRNSGLS | 1.718 ± 0.032 | 0.098 ± 0.011 |
| FE-341 motif | WP—-l | |||
| Rituximab motif | WP-WLE | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favoino, E.; Prete, M.; Catacchio, G.; Conteduca, G.; Perosa, F. CD20-Mimotope Peptides: A Model to Define the Molecular Basis of Epitope Spreading. Int. J. Mol. Sci. 2019, 20, 1920. https://doi.org/10.3390/ijms20081920
Favoino E, Prete M, Catacchio G, Conteduca G, Perosa F. CD20-Mimotope Peptides: A Model to Define the Molecular Basis of Epitope Spreading. International Journal of Molecular Sciences. 2019; 20(8):1920. https://doi.org/10.3390/ijms20081920
Chicago/Turabian StyleFavoino, Elvira, Marcella Prete, Giacomo Catacchio, Giuseppina Conteduca, and Federico Perosa. 2019. "CD20-Mimotope Peptides: A Model to Define the Molecular Basis of Epitope Spreading" International Journal of Molecular Sciences 20, no. 8: 1920. https://doi.org/10.3390/ijms20081920
APA StyleFavoino, E., Prete, M., Catacchio, G., Conteduca, G., & Perosa, F. (2019). CD20-Mimotope Peptides: A Model to Define the Molecular Basis of Epitope Spreading. International Journal of Molecular Sciences, 20(8), 1920. https://doi.org/10.3390/ijms20081920