Precision Revisited: Targeting Microcephaly Kinases in Brain Tumors



Abstract
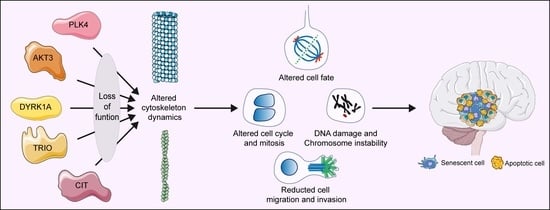
:
{kind=link}
{kind=link}
1. Background
1.1. The Hurdles of High-Grade Brain Tumor Precision Medicine
1.2. Congenital Microcephaly: A Tissue-Specific Phenotype of Ubiquitously Expressed Genes
1.3. Citron Kinase (CITK)
1.4. Polo-Like Kinase 4 (PLK4)
1.5. AKT Serine/Threonine Kinase 3 (AKT3)
1.6. Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1A (DYRK1A)
1.7. Trio Rho Guanine Nucleotide Exchange Factor (TRIO)
2. Remarks and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. (Berl.) 2012, 123, 465–472. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. (Berl.) 2016, 131, 821–831. [Google Scholar] [CrossRef]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Remke, M.; Hielscher, T.; Northcott, P.A.; Witt, H.; Ryzhova, M.; Wittmann, A.; Benner, A.; von Deimling, A.; Scheurlen, W.; Perry, A.; et al. Adult medulloblastoma comprises three major molecular variants. J. Clin. Oncol. 2011, 29, 2717–2723. [Google Scholar]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Penas-Prado, M.; Zhou, S.; Wei, J.; Khatua, S.; Hodges, T.R.; Sanai, N.; Xiu, J.; Gatalica, Z.; Kim, L.; et al. Rethinking medulloblastoma from a targeted therapeutics perspective. J. Neurooncol. 2018, 139, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, T.; Suresh, P.S. Emerging roles of MCPH1: Expedition from primary microcephaly to cancer. Eur. J. Cell Biol. 2014, 93, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, B.; Carlson, M.; Lu, K.V.; Zhu, S.; Felciano, R.M.; Laurance, M.F.; Zhao, W.; Qi, S.; Chen, Z.; et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl. Acad. Sci. USA 2006, 103, 17402–17407. [Google Scholar]
- Lang, P.Y.; Gershon, T.R. A New Way to Treat Brain Tumors: Targeting Proteins Coded by Microcephaly Genes?: Brain tumors and microcephaly arise from opposing derangements regulating progenitor growth. Drivers of microcephaly could be attractive brain tumor targets. BioEssays 2018, 40, e1700243. [Google Scholar]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar]
- Smith, A.W.; Mehta, M.P.; Wernicke, A.G. Neural stem cells, the subventricular zone and radiotherapy: Implications for treating glioblastoma. J. Neurooncol. 2016, 128, 207–216. [Google Scholar]
- Passemard, S.; Kaindl, A.M.; Verloes, A. Microcephaly. Handb. Clin. Neurol. 2013, 111, 129–141. [Google Scholar]
- Woods, C.G.; Parker, A. Investigating microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef]
- Zaqout, S.; Morris-Rosendahl, D.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly (MCPH): An Update. Neuropediatrics 2017, 48, 135–142. [Google Scholar]
- Abuelo, D. Microcephaly syndromes. Semin. Pediatr. Neurol. 2007, 14, 118–127. [Google Scholar] [CrossRef]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genomics 2015, 8 (Suppl. 1), S4. [Google Scholar] [CrossRef]
- O’Neill, R.S.; Schoborg, T.A.; Rusan, N.M. Same but different: Pleiotropy in centrosome-related microcephaly. Mol. Biol. Cell 2018, 29, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.T.; Tocco, C.; Pallavicini, G.; Liu, Y.; Vernì, F.; Merigliano, C.; Bonaccorsi, S.; El-Assawy, N.; Priano, L.; Gai, M.; et al. Citron Kinase Deficiency Leads to Chromosomal Instability and TP53-Sensitive Microcephaly. Cell Rep. 2017, 18, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Garcia, I.; Crowther, A.J.; Li, S.; Stewart, A.; Liu, H.; Lough, K.J.; O’Neill, S.; Veleta, K.; Oyarzabal, E.A.; et al. Aspm sustains postnatal cerebellar neurogenesis and medulloblastoma growth in mice. Dev. Camb. Engl. 2015, 142, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-W.; Tapias, A.; Bruhn, C.; Gruber, R.; Sukchev, M.; Wang, Z.-Q. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair 2013, 12, 645–655. [Google Scholar] [CrossRef] [PubMed]
- El-Ghouzzi, V.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly. Cell Death Dis. 2016, 7, e2440. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Q.; Jiang, Y.; Ye, Q.; Xu, D.; Gao, F.; Xu, J.W.; Wang, R.; Zhu, X.; Shi, L.; et al. Disruption of glial cell development by Zika virus contributes to severe microcephalic newborn mice. Cell Discov. 2018, 4, 43. [Google Scholar] [CrossRef]
- Lubin, J.A.; Zhang, R.R.; Kuo, J.S. Zika Virus has Oncolytic Activity Against Glioblastoma Stem Cells. Neurosurgery 2018, 82, E113–E114. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Neuro-oncology: A new role for Zika virus in glioblastoma therapy? Nat. Rev. Neurol. 2017, 13, 640–641. [Google Scholar] [CrossRef]
- Bikeye, S.-N.N.; Colin, C.; Marie, Y.; Vampouille, R.; Ravassard, P.; Rousseau, A.; Boisselier, B.; Idbaih, A.; Calvo, C.F.; Leuraud, P.; et al. ASPM-associated stem cell proliferation is involved in malignant progression of gliomas and constitutes an attractive therapeutic target. Cancer Cell Int. 2010, 10, 1. [Google Scholar] [CrossRef]
- Kato, T.A.; Okayasu, R.; Jeggo, P.A.; Fujimori, A. ASPM influences DNA double-strand break repair and represents a potential target for radiotherapy. Int. J. Radiat. Biol. 2011, 87, 1189–1195. [Google Scholar] [CrossRef]
- Huang, W.; Wang, J.; Zhang, D.; Chen, W.; Hou, L.; Wu, X.; Lu, Y. Inhibition of KIF14 Suppresses Tumor Cell Growth and Promotes Apoptosis in Human Glioblastoma. Cell. Physiol. Biochem. 2015, 37, 1659–1670. [Google Scholar] [CrossRef]
- Li, K.K.-W.; Qi, Y.; Xia, T.; Chan, A.K.-Y.; Zhang, Z.-Y.; Aibaidula, A.; Zhang, R.; Zhou, L.; Yao, Y.; Ng, H.-K. The kinesin KIF14 is overexpressed in medulloblastoma and downregulation of KIF14 suppressed tumor proliferation and induced apoptosis. Lab. Investig. 2017, 97, 946–961. [Google Scholar] [CrossRef]
- Li, M.; Xiao, A.; Floyd, D.; Olmez, I.; Lee, J.; Godlewski, J.; Bronisz, A.; Bhat, K.P.L.; Sulman, E.P.; Nakano, I.; et al. CDK4/6 inhibition is more active against the glioblastoma proneural subtype. Oncotarget 2017, 8, 55319–55331. [Google Scholar] [CrossRef]
- Yin, L.; Li, H.; Liu, W.; Yao, Z.; Cheng, Z.; Zhang, H.; Zou, H. A highly potent CDK4/6 inhibitor was rationally designed to overcome blood brain barrier in gliobastoma therapy. Eur. J. Med. Chem. 2018, 144, 1–28. [Google Scholar] [CrossRef]
- Di Cunto, F.; Calautti, E.; Hsiao, J.; Ong, L.; Topley, G.; Turco, E.; Dotto, G.P. Citron rho-interacting kinase, a novel tissue-specific ser/thr kinase encompassing the Rho-Rac-binding protein Citron. J. Biol. Chem. 1998, 273, 29706–29711. [Google Scholar] [CrossRef]
- Madaule, P.; Eda, M.; Watanabe, N.; Fujisawa, K.; Matsuoka, T.; Bito, H.; Ishizaki, T.; Narumiya, S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature 1998, 394, 491–494. [Google Scholar] [CrossRef]
- Naim, V.; Imarisio, S.; Di Cunto, F.; Gatti, M.; Bonaccorsi, S. Drosophila citron kinase is required for the final steps of cytokinesis. Mol. Biol. Cell 2004, 15, 5053–5063. [Google Scholar] [CrossRef]
- Liu, H.; Di Cunto, F.; Imarisio, S.; Reid, L.M. Citron kinase is a cell cycle-dependent, nuclear protein required for G2/M transition of hepatocytes. J. Biol. Chem. 2003, 278, 2541–2548. [Google Scholar] [CrossRef]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.M.; Annaratone, L.; et al. ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO Rep. 2016, 17, 1396–1409. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Audusseau, M.; Riparbelli, M.G.; Callaini, G.; D’Avino, P.P. Citron kinase controls a molecular network required for midbody formation in cytokinesis. Proc. Natl. Acad. Sci. USA 2013, 110, 9782–9787. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Verbrugghe, K.J.; Capalbo, L.; Gregory, S.; Montembault, E.; Glover, D.M.; D’Avino, P.P. Sticky/Citron kinase maintains proper RhoA localization at the cleavage site during cytokinesis. J. Cell Biol. 2011, 195, 595–603. [Google Scholar] [CrossRef]
- Dema, A.; Macaluso, F.; Sgrò, F.; Berto, G.E.; Bianchi, F.T.; Chiotto, A.A.; Pallavicini, G.; Di Cunto, F.; Gai, M. Citron kinase-dependent F-actin maintenance at midbody secondary ingression sites mediates abscission. J. Cell Sci. 2018, 131, jcs209080. [Google Scholar] [CrossRef]
- McKenzie, C.; Bassi, Z.I.; Debski, J.; Gottardo, M.; Callaini, G.; Dadlez, M.; D’Avino, P.P. Cross-regulation between Aurora B and Citron kinase controls midbody architecture in cytokinesis. Open Biol. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Sgrò, F.; Bianchi, F.T.; Falcone, M.; Pallavicini, G.; Gai, M.; Chiotto, A.M.A.; Berto, G.E.; Turco, E.; Chang, Y.J.; Huttner, W.B.; et al. Tissue-specific control of midbody microtubule stability by Citron kinase through modulation of TUBB3 phosphorylation. Cell Death Differ. 2016, 23, 801–813. [Google Scholar] [CrossRef]
- Di Cunto, F.; Imarisio, S.; Hirsch, E.; Broccoli, V.; Bulfone, A.; Migheli, A.; Atzori, C.; Turco, E.; Triolo, R.; Dotto, G.P.; et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 2000, 28, 115–127. [Google Scholar] [CrossRef]
- Sarkisian, M.R.; Li, W.; Di Cunto, F.; D’Mello, S.R.; LoTurco, J.J. Citron-kinase, a protein essential to cytokinesis in neuronal progenitors, is deleted in the flathead mutant rat. J. Neurosci. 2002, 22, RC217. [Google Scholar] [CrossRef]
- Di Cunto, F.; Imarisio, S.; Camera, P.; Boitani, C.; Altruda, F.; Silengo, L. Essential role of citron kinase in cytokinesis of spermatogenic precursors. J. Cell Sci. 2002, 115, 4819–4826. [Google Scholar] [CrossRef]
- Harding, B.N.; Moccia, A.; Drunat, S.; Soukarieh, O.; Tubeuf, H.; Chitty, L.S.; Verloes, A.; Gressens, P.; El Ghouzzi, V.; Joriot, S.; et al. Mutations in Citron Kinase Cause Recessive Microlissencephaly with Multinucleated Neurons. Am. J. Hum. Genet. 2016, 99, 511–520. [Google Scholar] [CrossRef]
- Li, H.; Bielas, S.L.; Zaki, M.S.; Ismail, S.; Farfara, D.; Um, K.; Rosti, R.O.; Scott, E.C.; Tu, S.; Chi, N.C.; et al. Biallelic Mutations in Citron Kinase Link Mitotic Cytokinesis to Human Primary Microcephaly. Am. J. Hum. Genet. 2016, 99, 501–510. [Google Scholar] [CrossRef]
- Basit, S.; Al-Harbi, K.M.; Alhijji, S.A.M.; Albalawi, A.M.; Alharby, E.; Eldardear, A.; Samman, M.I. CIT, a gene involved in neurogenic cytokinesis, is mutated in human primary microcephaly. Hum. Genet. 2016, 135, 1199–1207. [Google Scholar] [CrossRef]
- Shaheen, R.; Hashem, A.; Abdel-Salam, G.M.H.; Al-Fadhli, F.; Ewida, N.; Alkuraya, F.S. Mutations in CIT, encoding citron rho-interacting serine/threonine kinase, cause severe primary microcephaly in humans. Hum. Genet. 2016, 135, 1191–1197. [Google Scholar] [CrossRef]
- D’Avino, P.P. Citron kinase - renaissance of a neglected mitotic kinase. J. Cell Sci. 2017, 130, 1701–1708. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, J.; Wang, K.-S.; Zhang, X.; Han, Z.-G. RNA interference targeting CITRON can significantly inhibit the proliferation of hepatocellular carcinoma cells. Mol. Biol. Rep. 2011, 38, 693–702. [Google Scholar] [CrossRef]
- Ehrlichova, M.; Mohelnikova-Duchonova, B.; Hrdy, J.; Brynychova, V.; Mrhalova, M.; Kodet, R.; Rob, L.; Pluta, M.; Gut, I.; Soucek, P.; et al. The association of taxane resistance genes with the clinical course of ovarian carcinoma. Genomics 2013, 102, 96–101. [Google Scholar] [CrossRef]
- Tong, H.; Wang, J.; Chen, H.; Wang, Z.; Fan, H.; Ni, Z. Transcriptomic analysis of gene expression profiles of stomach carcinoma reveal abnormal expression of mitotic components. Life Sci. 2017, 170, 41–49. [Google Scholar] [CrossRef]
- Gruneberg, U.; Neef, R.; Li, X.; Chan, E.H.Y.; Chalamalasetty, R.B.; Nigg, E.A.; Barr, F.A. KIF14 and citron kinase act together to promote efficient cytokinesis. J. Cell Biol. 2006, 172, 363–372. [Google Scholar] [CrossRef]
- McKenzie, C.; D’Avino, P.P. Investigating cytokinesis failure as a strategy in cancer therapy. Oncotarget 2016, 7, 87323–87341. [Google Scholar] [CrossRef]
- Pallavicini, G.; Sgrò, F.; Garello, F.; Falcone, M.; Bitonto, V.; Berto, G.E.; Bianchi, F.T.; Gai, M.; Chiotto, A.M.A.; Filippi, M.; et al. Inactivation of Citron Kinase Inhibits Medulloblastoma Progression by Inducing Apoptosis and Cell Senescence. Cancer Res. 2018, 78, 4599–4612. [Google Scholar] [CrossRef]
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and function of Polo-like kinases. Oncogene 2005, 24, 248–259. [Google Scholar] [CrossRef]
- Maniswami, R.R.; Prashanth, S.; Karanth, A.V.; Koushik, S.; Govindaraj, H.; Mullangi, R.; Rajagopal, S.; Jegatheesan, S.K. PLK4: A link between centriole biogenesis and cancer. Expert Opin. Ther. Targets 2018, 22, 59–73. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef]
- Karn, T.; Holtrich, U.; Wolf, G.; Hock, B.; Strebhardt, K.; Rubsamenwaigmann, H. Human SAK related to the PLK/polo family of cell cycle kinases shows high mRNA expression in testis. Oncol. Rep. 1997, 4, 505–510. [Google Scholar] [CrossRef]
- Sillibourne, J.E.; Tack, F.; Vloemans, N.; Boeckx, A.; Thambirajah, S.; Bonnet, P.; Ramaekers, F.C.S.; Bornens, M.; Grand-Perret, T. Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 2010, 21, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.W.; Kozarova, A.; Cheung, P.; Macmillan, J.C.; Swallow, C.J.; Cross, J.C.; Dennis, J.W. Late mitotic failure in mice lacking Sak, a polo-like kinase. Curr. Biol. 2001, 11, 441–446. [Google Scholar] [CrossRef]
- Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Habedanck, R.; Stierhof, Y.-D.; Nigg, E.A. Plk4-induced centriole biogenesis in human cells. Dev. Cell 2007, 13, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Rodrigues-Martins, A.; Carpenter, L.; Riparbelli, M.; Lehmann, L.; Gatt, M.K.; Carmo, N.; Balloux, F.; Callaini, G.; Glover, D.M. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 2005, 15, 2199–2207. [Google Scholar] [CrossRef]
- Duensing, A.; Liu, Y.; Perdreau, S.A.; Kleylein-Sohn, J.; Nigg, E.A.; Duensing, S. Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene 2007, 26, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Habedanck, R.; Stierhof, Y.-D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-S.; Park, J.-E.; Shukla, A.; Choi, S.; Murugan, R.N.; Lee, J.H.; Ahn, M.; Rhee, K.; Bang, J.K.; Kim, B.Y.; et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc. Natl. Acad. Sci. USA 2013, 110, E4849–E4857. [Google Scholar] [CrossRef]
- Sonnen, K.F.; Gabryjonczyk, A.-M.; Anselm, E.; Stierhof, Y.-D.; Nigg, E.A. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J. Cell Sci. 2013, 126, 3223–3233. [Google Scholar] [CrossRef]
- Kratz, A.-S.; Bärenz, F.; Richter, K.T.; Hoffmann, I. Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol. Open 2015, 4, 370–377. [Google Scholar] [CrossRef]
- Ohta, M.; Ashikawa, T.; Nozaki, Y.; Kozuka-Hata, H.; Goto, H.; Inagaki, M.; Oyama, M.; Kitagawa, D. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat. Commun. 2014, 5, 5267. [Google Scholar] [CrossRef]
- Petrinac, S.; Ganuelas, M.L.; Bonni, S.; Nantais, J.; Hudson, J.W. Polo-like kinase 4 phosphorylates Chk2. Cell Cycle 2009, 8, 327–329. [Google Scholar] [CrossRef]
- Rosario, C.O.; Ko, M.A.; Haffani, Y.Z.; Gladdy, R.A.; Paderova, J.; Pollett, A.; Squire, J.A.; Dennis, J.W.; Swallow, C.J. Plk4 is required for cytokinesis and maintenance of chromosomal stability. Proc. Natl. Acad. Sci. USA 2010, 107, 6888–6893. [Google Scholar] [CrossRef]
- Holland, A.J.; Fachinetti, D.; Da Cruz, S.; Zhu, Q.; Vitre, B.; Lince-Faria, M.; Chen, D.; Parish, N.; Verma, I.M.; Bettencourt-Dias, M.; et al. Polo-like kinase 4 controls centriole duplication but does not directly regulate cytokinesis. Mol. Biol. Cell 2012, 23, 1838–1845. [Google Scholar] [CrossRef]
- Martin, C.-A.; Ahmad, I.; Klingseisen, A.; Hussain, M.S.; Bicknell, L.S.; Leitch, A.; Nürnberg, G.; Toliat, M.R.; Murray, J.E.; Hunt, D.; et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat. Genet. 2014, 46, 1283–1292. [Google Scholar] [CrossRef]
- Dinçer, T.; Yorgancıoğlu-Budak, G.; Ölmez, A.; Er, İ.; Dodurga, Y.; Özdemir, Ö.M.; Toraman, B.; Yıldırım, A.; Sabir, N.; Akarsu, N.A.; et al. Analysis of centrosome and DNA damage response in PLK4 associated Seckel syndrome. Eur. J. Hum. Genet. 2017, 25, 1118–1125. [Google Scholar] [CrossRef]
- Ko, M.A.; Rosario, C.O.; Hudson, J.W.; Kulkarni, S.; Pollett, A.; Dennis, J.W.; Swallow, C.J. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nat. Genet. 2005, 37, 883–888. [Google Scholar] [CrossRef]
- Li, Z.; Dai, K.; Wang, C.; Song, Y.; Gu, F.; Liu, F.; Fu, L. Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J. Cancer 2016, 7, 1125–1132. [Google Scholar] [CrossRef]
- Kazazian, K.; Go, C.; Wu, H.; Brashavitskaya, O.; Xu, R.; Dennis, J.W.; Gingras, A.-C.; Swallow, C.J. Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res. 2017, 77, 434–447. [Google Scholar] [CrossRef]
- Mason, J.M.; Lin, D.C.-C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef]
- Lohse, I.; Mason, J.; Cao, P.M.; Pintilie, M.; Bray, M.; Hedley, D.W. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017, 8, 3064–3071. [Google Scholar] [CrossRef]
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 1913–1918. [Google Scholar] [CrossRef]
- Oegema, K.; Davis, R.L.; Lara-Gonzalez, P.; Desai, A.; Shiau, A.K. CFI-400945 is not a selective cellular PLK4 inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, E10808–E10809. [Google Scholar] [CrossRef]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef]
- Meitinger, F.; Anzola, J.V.; Kaulich, M.; Richardson, A.; Stender, J.D.; Benner, C.; Glass, C.K.; Dowdy, S.F.; Desai, A.; Shiau, A.K.; et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J. Cell Biol. 2016, 214, 155–166. [Google Scholar] [CrossRef]
- Sredni, S.T.; Bailey, A.W.; Suri, A.; Hashizume, R.; He, X.; Louis, N.; Gokirmak, T.; Piper, D.R.; Watterson, D.M.; Tomita, T. Inhibition of polo-like kinase 4 (PLK4): A new therapeutic option for rhabdoid tumors and pediatric medulloblastoma. Oncotarget 2017, 8, 111190–111212. [Google Scholar] [CrossRef]
- Sredni, S.T.; Tomita, T. The polo-like kinase 4 gene (PLK4) is overexpressed in pediatric medulloblastoma. Childs Nerv. Syst. 2017, 33, 1031. [Google Scholar] [CrossRef]
- Bailey, A.W.; Suri, A.; Chou, P.M.; Pundy, T.; Gadd, S.; Raimondi, S.L.; Tomita, T.; Sredni, S.T. Polo-Like Kinase 4 (PLK4) Is Overexpressed in Central Nervous System Neuroblastoma (CNS-NB). Bioengineering 2018, 5, 96. [Google Scholar] [CrossRef]
- Sredni, S.T.; Suzuki, M.; Yang, J.-P.; Topczewski, J.; Bailey, A.W.; Gokirmak, T.; Gross, J.N.; de Andrade, A.; Kondo, A.; Piper, D.R.; et al. A functional screening of the kinome identifies the Polo-like kinase 4 as a potential therapeutic target for malignant rhabdoid tumors, and possibly, other embryonal tumors of the brain. Pediatr. Blood Cancer 2017, 64, e26551. [Google Scholar] [CrossRef]
- Wang, J.; Zuo, J.; Wang, M.; Ma, X.; Gao, K.; Bai, X.; Wang, N.; Xie, W.; Liu, H. Polo-like kinase 4 promotes tumorigenesis and induces resistance to radiotherapy in glioblastoma. Oncol. Rep. 2019, 41, 2159–2167. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Huang, K.; Liu, Y.; Wei, C.; Zhou, J.; Zhang, W.; Wang, Q.; Liang, H.; Zhang, A.; et al. PLK4 is a determinant of temozolomide sensitivity through phosphorylation of IKBKE in glioblastoma. Cancer Lett. 2019, 443, 91–107. [Google Scholar] [CrossRef]
- Fayard, E.; Tintignac, L.A.; Baudry, A.; Hemmings, B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim. Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Tschopp, O.; Baudry, A.; Dümmler, B.; Hynx, D.; Hemmings, B.A. Physiological functions of protein kinase B/Akt. Biochem. Soc. Trans. 2004, 32, 350–354. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis Int. J. Program. Cell Death 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Brodbeck, D.; Cron, P.; Hemmings, B.A. A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J. Biol. Chem. 1999, 274, 9133–9136. [Google Scholar] [CrossRef]
- Masure, S.; Haefner, B.; Wesselink, J.J.; Hoefnagel, E.; Mortier, E.; Verhasselt, P.; Tuytelaars, A.; Gordon, R.; Richardson, A. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. Eur. J. Biochem. 1999, 265, 353–360. [Google Scholar] [CrossRef]
- Poduri, A.; Evrony, G.D.; Cai, X.; Elhosary, P.C.; Beroukhim, R.; Lehtinen, M.K.; Hills, L.B.; Heinzen, E.L.; Hill, A.; Hill, R.S.; et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012, 74, 41–48. [Google Scholar] [CrossRef]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.-Y.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef]
- Tschopp, O.; Yang, Z.-Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Dev. Camb. Engl. 2005, 132, 2943–2954. [Google Scholar]
- Boland, E.; Clayton-Smith, J.; Woo, V.G.; McKee, S.; Manson, F.D.C.; Medne, L.; Zackai, E.; Swanson, E.A.; Fitzpatrick, D.; Millen, K.J.; et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am. J. Hum. Genet. 2007, 81, 292–303. [Google Scholar] [CrossRef]
- Rivière, J.-B.; Mirzaa, G.M.; O’Roak, B.J.; Beddaoui, M.; Alcantara, D.; Conway, R.L.; St-Onge, J.; Schwartzentruber, J.A.; Gripp, K.W.; Nikkel, S.M.; et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012, 44, 934–940. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef]
- Toker, A. Achieving specificity in Akt signaling in cancer. Adv. Biol. Regul. 2012, 52, 78–87. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef]
- Ichimura, K.; Vogazianou, A.P.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Plant, K.; Baird, K.; Langford, C.F.; Gregory, S.G.; Collins, V.P. 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumours and homozygously deleted in a subset of glioblastomas. Oncogene 2008, 27, 2097–2108. [Google Scholar] [CrossRef]
- Kagawa, N.; Maruno, M.; Suzuki, T.; Hashiba, T.; Hashimoto, N.; Izumoto, S.; Yoshimine, T. Detection of genetic and chromosomal aberrations in medulloblastomas and primitive neuroectodermal tumors with DNA microarrays. Brain Tumor Pathol. 2006, 23, 41–47. [Google Scholar] [CrossRef]
- Endersby, R.; Zhu, X.; Hay, N.; Ellison, D.W.; Baker, S.J. Nonredundant functions for Akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res. 2011, 71, 4106–4116. [Google Scholar] [CrossRef]
- Paul-Samojedny, M.; Pudełko, A.; Suchanek-Raif, R.; Kowalczyk, M.; Fila-Daniłow, A.; Borkowska, P.; Kowalski, J. Knockdown of the AKT3 (PKBγ), PI3KCA, and VEGFR2 genes by RNA interference suppresses glioblastoma multiforme T98G cells invasiveness in vitro. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 3263–3277. [Google Scholar] [CrossRef]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef]
- Paul-Samojedny, M.; Pudełko, A.; Kowalczyk, M.; Fila-Daniłow, A.; Suchanek-Raif, R.; Borkowska, P.; Kowalski, J. Combination Therapy with AKT3 and PI3KCA siRNA Enhances the Antitumor Effect of Temozolomide and Carmustine in T98G Glioblastoma Multiforme Cells. BioDrugs 2016, 30, 129–144. [Google Scholar] [CrossRef]
- Becker, W.; Joost, H.G. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid Res. Mol. Biol. 1999, 62, 1–17. [Google Scholar]
- Kay, L.J.; Smulders-Srinivasan, T.K.; Soundararajan, M. Understanding the Multifaceted Role of Human Down Syndrome Kinase DYRK1A. Adv. Protein Chem. Struct. Biol. 2016, 105, 127–171. [Google Scholar]
- Galceran, J.; de Graaf, K.; Tejedor, F.J.; Becker, W. The MNB/DYRK1A protein kinase: Genetic and biochemical properties. J. Neural Transm. Suppl. 2003, 67, 139–148. [Google Scholar]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef]
- Song, W.J.; Sternberg, L.R.; Kasten-Sportès, C.; Keuren, M.L.; Chung, S.H.; Slack, A.C.; Miller, D.E.; Glover, T.W.; Chiang, P.W.; Lou, L.; et al. Isolation of human and murine homologues of the Drosophila minibrain gene: Human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics 1996, 38, 331–339. [Google Scholar]
- Fotaki, V.; Dierssen, M.; Alcántara, S.; Martínez, S.; Martí, E.; Casas, C.; Visa, J.; Soriano, E.; Estivill, X.; Arbonés, M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002, 22, 6636–6647. [Google Scholar] [CrossRef]
- Van Bon, B.W.M.; Hoischen, A.; Hehir-Kwa, J.; de Brouwer, A.P.M.; Ruivenkamp, C.; Gijsbers, A.C.J.; Marcelis, C.L.; de Leeuw, N.; Veltman, J.A.; Brunner, H.G.; et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 2011, 79, 296–299. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef]
- Bellmaine, S.F.; Ovchinnikov, D.A.; Manallack, D.T.; Cuddy, C.E.; Elefanty, A.G.; Stanley, E.G.; Wolvetang, E.J.; Williams, S.J.; Pera, M. Inhibition of DYRK1A disrupts neural lineage specificationin human pluripotent stem cells. eLife 2017, 6, e24502. [Google Scholar] [CrossRef]
- Kurabayashi, N.; Nguyen, M.D.; Sanada, K. Triple play of DYRK1A kinase in cortical progenitor cells of Trisomy 21. Neurosci. Res. 2019, 138, 19–25. [Google Scholar] [CrossRef]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef]
- Laguna, A.; Aranda, S.; Barallobre, M.J.; Barhoum, R.; Fernández, E.; Fotaki, V.; Delabar, J.M.; de la Luna, S.; de la Villa, P.; Arbonés, M.L. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 2008, 15, 841–853. [Google Scholar] [CrossRef]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.-P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Gwack, Y.; Sharma, S.; Nardone, J.; Tanasa, B.; Iuga, A.; Srikanth, S.; Okamura, H.; Bolton, D.; Feske, S.; Hogan, P.G.; et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 2006, 441, 646–650. [Google Scholar] [CrossRef]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef]
- Ori-McKenney, K.M.; McKenney, R.J.; Huang, H.H.; Li, T.; Meltzer, S.; Jan, L.Y.; Vale, R.D.; Wiita, A.P.; Jan, Y.N. Phosphorylation of β-Tubulin by the Down Syndrome Kinase, Minibrain/DYRK1a, Regulates Microtubule Dynamics and Dendrite Morphogenesis. Neuron 2016, 90, 551–563. [Google Scholar] [CrossRef]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef]
- Pozo, N.; Zahonero, C.; Fernández, P.; Liñares, J.M.; Ayuso, A.; Hagiwara, M.; Pérez, A.; Ricoy, J.R.; Hernández-Laín, A.; Sepúlveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Invest. 2013, 123, 2475–2487. [Google Scholar] [CrossRef]
- Zhou, Q.; Phoa, A.F.; Abbassi, R.H.; Hoque, M.; Reekie, T.A.; Font, J.S.; Ryan, R.M.; Stringer, B.W.; Day, B.W.; Johns, T.G.; et al. Structural Optimization and Pharmacological Evaluation of Inhibitors Targeting Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases (DYRK) and CDC-like kinases (CLK) in Glioblastoma. J. Med. Chem. 2017, 60, 2052–2070. [Google Scholar] [CrossRef]
- Shimokawa, T.; Rahman, M.F.-U.; Tostar, U.; Sonkoly, E.; Ståhle, M.; Pivarcsi, A.; Palaniswamy, R.; Zaphiropoulos, P.G. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013, 10, 321–333. [Google Scholar] [CrossRef]
- Debant, A.; Serra-Pagès, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e29769. [Google Scholar] [CrossRef]
- Blangy, A.; Vignal, E.; Schmidt, S.; Debant, A.; Gauthier-Rouvière, C.; Fort, P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J. Cell Sci. 2000, 113, 729–739. [Google Scholar]
- Medley, Q.G.; Buchbinder, E.G.; Tachibana, K.; Ngo, H.; Serra-Pagès, C.; Streuli, M. Signaling between focal adhesion kinase and trio. J. Biol. Chem. 2003, 278, 13265–13270. [Google Scholar] [CrossRef]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef]
- Varvagiannis, K.; Vissers, L.E.; Baralle, D.; de Vries, B.B. TRIO-Related Intellectual Disability. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Ba, W.; Yan, Y.; Reijnders, M.R.F.; Schuurs-Hoeijmakers, J.H.M.; Feenstra, I.; Bongers, E.M.H.F.; Bosch, D.G.M.; De Leeuw, N.; Pfundt, R.; Gilissen, C.; et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2016, 25, 892–902. [Google Scholar] [CrossRef]
- Estrach, S.; Schmidt, S.; Diriong, S.; Penna, A.; Blangy, A.; Fort, P.; Debant, A. The Human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr. Biol. CB 2002, 12, 307–312. [Google Scholar] [CrossRef]
- DeGeer, J.; Boudeau, J.; Schmidt, S.; Bedford, F.; Lamarche-Vane, N.; Debant, A. Tyrosine phosphorylation of the Rho guanine nucleotide exchange factor Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol. Cell. Biol. 2013, 33, 739–751. [Google Scholar] [CrossRef]
- Fortin, S.P.; Ennis, M.J.; Schumacher, C.A.; Zylstra-Diegel, C.R.; Williams, B.O.; Ross, J.T.D.; Winkles, J.A.; Loftus, J.C.; Symons, M.H.; Tran, N.L. Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol. Cancer Res. MCR 2012, 10, 958–968. [Google Scholar] [CrossRef]
- Salhia, B.; Tran, N.L.; Chan, A.; Wolf, A.; Nakada, M.; Rutka, F.; Ennis, M.; McDonough, W.S.; Berens, M.E.; Symons, M.; et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am. J. Pathol. 2008, 173, 1828–1838. [Google Scholar] [CrossRef]
- Bouquier, N.; Fromont, S.; Debant, A.; Schmidt, S. Random mutagenesis of peptide aptamers as an optimization strategy for inhibitor screening. Methods Mol. Biol. 2012, 928, 97–118. [Google Scholar]
- Yano, T.; Yamazaki, Y.; Adachi, M.; Okawa, K.; Fort, P.; Uji, M.; Tsukita, S.; Tsukita, S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J. Cell Biol. 2011, 193, 319–332. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallavicini, G.; Berto, G.E.; Di Cunto, F. Precision Revisited: Targeting Microcephaly Kinases in Brain Tumors. Int. J. Mol. Sci. 2019, 20, 2098. https://doi.org/10.3390/ijms20092098
Pallavicini G, Berto GE, Di Cunto F. Precision Revisited: Targeting Microcephaly Kinases in Brain Tumors. International Journal of Molecular Sciences. 2019; 20(9):2098. https://doi.org/10.3390/ijms20092098
Chicago/Turabian StylePallavicini, Gianmarco, Gaia E. Berto, and Ferdinando Di Cunto. 2019. "Precision Revisited: Targeting Microcephaly Kinases in Brain Tumors" International Journal of Molecular Sciences 20, no. 9: 2098. https://doi.org/10.3390/ijms20092098
APA StylePallavicini, G., Berto, G. E., & Di Cunto, F. (2019). Precision Revisited: Targeting Microcephaly Kinases in Brain Tumors. International Journal of Molecular Sciences, 20(9), 2098. https://doi.org/10.3390/ijms20092098