Multifaceted Functional Role of Semaphorins in Glioblastoma

Abstract
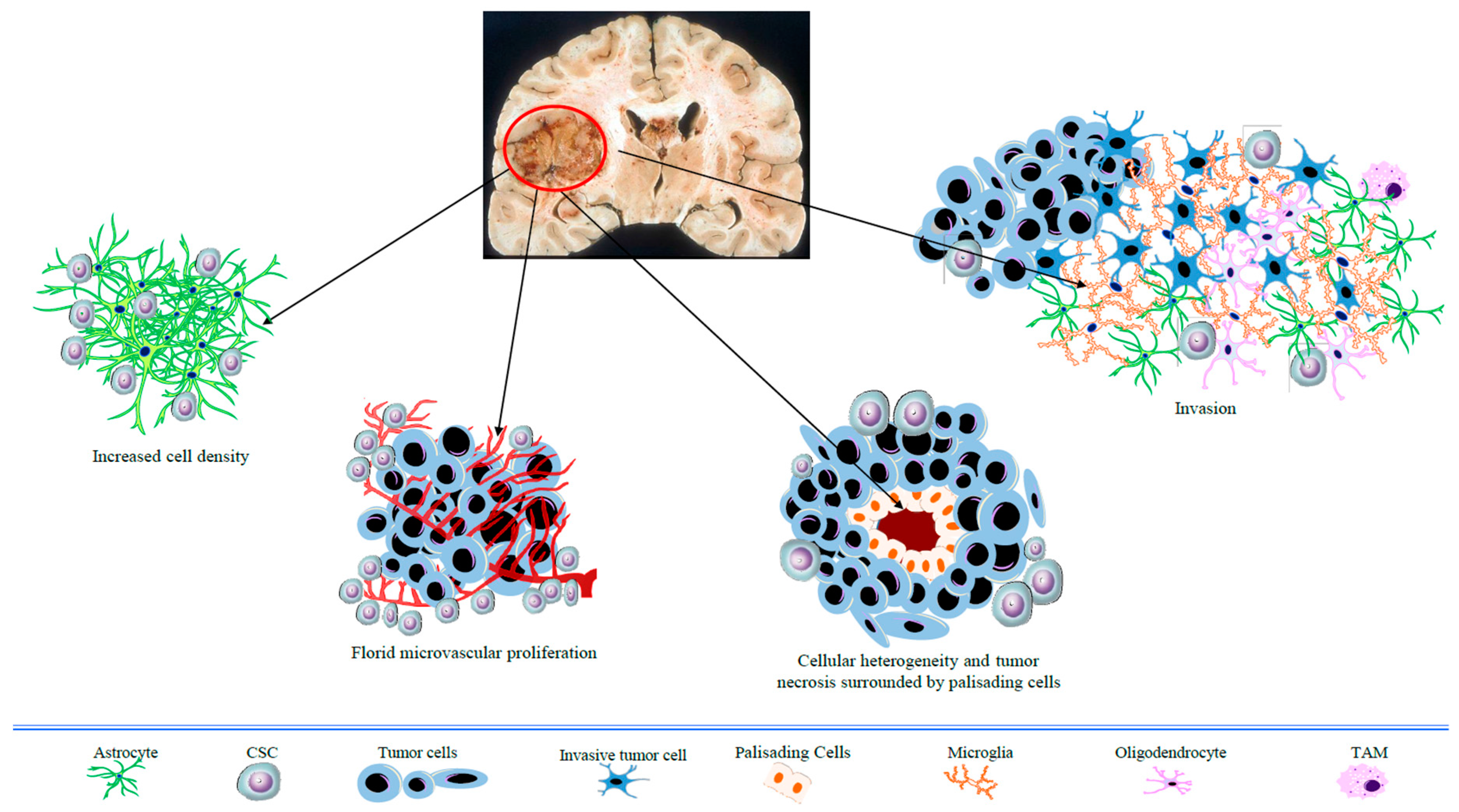
:1. Introduction
2. Role of Semaphorins in GBM Cell Growth and Survival
3. Role of Semaphorin Signaling in GBM Cell Migration and Invasiveness
4. Role of Semaphorins in GBM Angiogenesis
5. Role of Semaphorins in GBM Progression by Modulation of Immune Response
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Mangiola, A.; Lama, G.; Giannitelli, C.; De Bonis, P.; Anile, C.; Lauriola, L.; La Torre, G.; Sabatino, G.; Maira, G.; Jhanwar-Uniyal, M.; et al. Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme: Possible prognostic implications. Clin. Cancer. Res. 2007, 13, 6970–6977, Erratum in: Clin. Cancer. Res. 2008, 14, 4995–4996. [Google Scholar] [CrossRef]
- Lama, G.; Mangiola, A.; Anile, C.; Sabatino, G.; De Bonis, P.; Lauriola, L.; Giannitelli, C.; La Torre, G.; Jhanwar-Uniyal, M.; Sica, G.; et al. Activated ERK1/2 expression in glioblastoma multiforme and in peritumor tissue. Int. J. Oncol. 2007, 30, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Lama, G.; Mangiola, A.; Proietti, G.; Colabianchi, A.; Angelucci, C.; D’ Alessio, A.; De Bonis, P.; Geloso, M.C.; Lauriola, L.; Binda, E.; et al. Progenitor/Stem Cell Markers in Brain Adjacent to Glioblastoma: GD3 Ganglioside and NG2 Proteoglycan Expression. J. Neuropathol. Exp. Neurol. 2016, 75, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelucci, C.; D’Alessio, A.; Lama, G.; Binda, E.; Mangiola, A.; Vescovi, A.L.; Proietti, G.; Masuelli, L.; Bei, R.; Fazi, B.; et al. Cancer stem cells from peritumoral tissue of glioblastoma multiforme: The possible missing link between tumor development and progression. Oncotarget 2018, 9, 28116–28130. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Ferris, S.P.; Hofmann, J.W.; Solomon, D.A.; Perry, A. Characterization of gliomas: From morphology to molecules. Virchows Arch. 2017, 471, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qin, Z.; Chen, Z.; Xie, L.; Wang, R.; Zhao, H. Tumor Microenvironment in Treatment of Glioma. Open Med. (Wars) 2017, 12, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werbowetski, T.; Bjerkvig, R.; Del Maestro, R.F. Evidence for a secreted chemorepellent that directs glioma cell invasion. J. Neurobiol. 2004, 60, 71–88. [Google Scholar] [CrossRef] [Green Version]
- Jarjour, A.A.; Durko, M.; Luk, T.L.; Marçal, N.; Shekarabi, M.; Kennedy, T.E. Autocrine netrin function inhibits glioma cell motility and promotes focal adhesion formation. PLoS ONE 2011, 6, e25408. [Google Scholar] [CrossRef] [PubMed]
- Mertsch, S.; Schmitz, N.; Jeibmann, A.; Geng, J.G.; Paulus, W.; Senner, V. Slit2 involvement in glioma cell migration is mediated by Robo1 receptor. J. Neurooncol. 2008, 87, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin, A.L.; Matthes, D.J.; Goodman, C.S. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 1993, 75, 1389–1399. [Google Scholar] [CrossRef]
- Tamagnone, L.; Artigiani, S.; Chen, H.; He, Z.; Ming, G.I.; Song, H.; Chedotal, A.; Winberg, M.L.; Goodman, C.S.; Poo, M.; et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 1999, 99, 71–80, Erratum in: Cell 2001. [Google Scholar] [CrossRef]
- Roche, J.; Boldog, F.; Robinson, M.; Robinson, L.; Varella-Garcia, M.; Swanton, M.; Waggoner, B.; Fishel, R.; Franklin, W.; Gemmill, R.; et al. Distinct 3p21.3 deletions in lung cancer and identification of a new human semaphorin. Oncogene 1996, 12, 1289–1297. [Google Scholar]
- Xiang, R.H.; Hensel, C.H.; Garcia, D.K.; Carlson, H.C.; Kok, K.; Daly, M.C.; Kerbacher, K.; van denBerg, A.; Veldhuis, P.; Buys, C.H.; et al. Isolation of the human semaphorin III/F gene (SEMA3F) at chromosome 3p21, a region deleted in lung cancer. Genomics 1996, 32, 39–48. [Google Scholar] [CrossRef]
- Law, J.W.; Lee, A.Y. The role of semaphorins and their receptors in gliomas. J. Signal Transduct. 2012, 2012, 902854. [Google Scholar] [CrossRef]
- Sadanandam, A.; Sidhu, S.S.; Wullschleger, S.; Singh, S.; Varney, M.L.; Yang, C.S.; Ashour, A.E.; Batra, S.K.; Singh, R.K. Secreted semaphorin 5A suppressed pancreatic tumour burden but increased metastasis and endothelial cell proliferation. Br. J. Cancer 2012, 107, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Zhang, X.; Ren, J.; Lu, J.; Li, W.; Fu, H.; Zhang, S.; Li, J. Semaphorin 5A, an axon guidance molecule, enhances the invasion and metastasis of human gastric cancer through activation of MMP9. Pathol. Oncol. Res. 2013, 19, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, G.; Mumblat, Y.; Smolkin, T.; Toledano, S.; Nir-Zvi, I.; Ziv, K.; Kessler, O. The semaphorins and their receptors as modulators of tumor progression. Drug Resist. Updat. 2016, 29, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gurrapu, S.; Tamagnone, L. Transmembrane semaphorins: Multimodal signaling cues in development and cancer. Cell Adh. Migr. 2016, 10, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurrapu, S.; Pupo, E.; Franzolin, G.; Lanzetti, L.; Tamagnone, L. Sema4C/PlexinB2 signaling controls breast cancer cell growth, hormonal dependence and tumorigenic potential. Cell Death Differ. 2018, 25, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Rieger, J.; Wick, W.; Weller, M. Human malignant glioma cells express semaphorins and their receptors, neuropilins and plexins. Glia 2003, 42, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Sabag, A.D.; Bode, J.; Fink, D.; Kigel, B.; Kugler, W.; Neufeld, G. Semaphorin-3D and semaphorin-3E inhibit the development of tumors from glioblastoma cells implanted in the cortex of the brain. PLoS ONE 2012, 7, e42912. [Google Scholar] [CrossRef]
- Nasarre, C.; Koncina, E.; Labourdette, G.; Cremel, G.; Roussel, G.; Aunis, D.; Bagnard, D. Neuropilin-2 acts as a modulator of Sema3A-dependent glioma cell migration. Cell Adh. Migr. 2009, 3, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Shin, Y.J.; Lee, K.; Cho, H.J.; Sa, J.K.; Lee, S.Y.; Kim, S.H.; Lee, J.; Yoon, Y.; Nam, DH. Anti-SEMA3A Antibody: A Novel Therapeutic Agent to Suppress Glioblastoma Tumor Growth. Cancer Res. Treat. 2018, 50, 1009–1022. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.; Son, E.; Yoo, S.J.; Sa, J.K.; Shin, Y.J.; Yoon, Y.; Nam, D.H. Pharmacokinetics, Biodistribution, and Toxicity Evaluation of Anti-SEMA3A (F11) in In Vivo Models. Anticancer Res. 2018, 38, 2803–2810. [Google Scholar] [CrossRef]
- Bagci, T.; Wu, J.K.; Pfannl, R.; Ilag, L.L.; Jay, D.G. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene 2009, 28, 3537–3550. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Qiao, S.; Chen, K. Suppression of miR-221 inhibits glioma cells proliferation and invasion via targeting SEMA3B. Biol. Res. 2015, 48, 37. [Google Scholar] [CrossRef]
- Nakayama, H.; Bruneau, S.; Kochupurakkal, N.; Coma, S.; Briscoe, D.M.; Klagsbrun, M. Regulation of mTOR Signaling by Semaphorin 3F-Neuropilin 2 Interactions In Vitro and In Vivo. Sci. Rep. 2015, 5, 11789. [Google Scholar] [CrossRef] [Green Version]
- Man, J.; Shoemake, J.; Zhou, W.; Fang, X.; Wu, Q.; Rizzo, A.; Prayson, R.; Bao, S.; Rich, J.N.; Yu, J.S. Sema3C promotes the survival and tumorigenicity of glioma stem cells through Rac1 activation. Cell Rep. 2014, 9, 1812–1826. [Google Scholar] [CrossRef]
- Chang, Y.; Li, L.; Zhang, L.; Guo, X.; Feng, Z.; Zhou, J.; Zhou, S.; Feng, G.; Han, F.; Huang, W.; et al. Plexin-B1 indirectly affects glioma invasiveness and angiogenesis by regulating the RhoA/αvβ3 signaling pathway and SRPK1. Tumour Biol. 2016, 37, 11225–11236. [Google Scholar] [CrossRef]
- Giordano, S.; Corso, S.; Conrotto, P.; Artigiani, S.; Gilestro, G.; Barberis, D.; Tamagnone, L.; Comoglio, P.M. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 2002, 4, 720–724. [Google Scholar] [CrossRef]
- Hu, B.; Guo, P.; Bar-Joseph, I.; Imanishi, Y.; Jarzynka, M.J.; Bogler, O.; Mikkelsen, T.; Hirose, T.; Nishikawa, R.; Cheng, S.Y. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene 2007, 26, 5577–5586. [Google Scholar] [CrossRef] [Green Version]
- Kigel, B.; Rabinowicz, N.; Varshavsky, A.; Kessler, O.; Neufeld, G. Plexin-A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 2011, 118, 4285–4296. [Google Scholar] [CrossRef] [Green Version]
- Procaccia, V.; Nakayama, H.; Shimizu, A.; Klagsbrun, M. Gleevec/imatinib, an ABL2 kinase inhibitor, protects tumor and endothelial cells from semaphorin-induced cytoskeleton collapse and loss of cell motility. Biochem. Biophys. Res. Commun. 2014, 448, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, A.; Mammoto, A.; Italiano, J.E., Jr.; Pravda, E.; Dudley, A.C.; Ingber, D.E.; Klagsbrun, M. ABL2/ARG tyrosine kinase mediates SEMA3F-induced RhoA inactivation and cytoskeleton collapse in human glioma cells. J. Biol. Chem. 2008, 283, 27230–27238. [Google Scholar] [CrossRef]
- Coma, S.; Shimizu, A.; Klagsbrun, M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adh. Migr. 2011, 5, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Santra, M.; Zhang, X.; Santra, S.; Jiang, F.; Chopp, M. Ectopic doublecortin gene expression suppresses the malignant phenotype in glioblastoma cells. Cancer Res. 2006, 66, 11726–11735. [Google Scholar] [CrossRef]
- Zhou, X.; Ma, L.; Li, J.; Gu, J.; Shi, Q.; Yu, R. Effects of SEMA3G on migration and invasion of glioma cells. Oncol. Rep. 2012, 28, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Le, A.P.; Huang, Y.; Pingle, S.C.; Kesari, S.; Wang, H.; Yong, R.L.; Zou, H.; Friedel, R.H. Plexin-B2 promotes invasive growth of malignant glioma. Oncotarget 2015, 6, 7293–7304. [Google Scholar] [CrossRef] [Green Version]
- Artigiani, S.; Conrotto, P.; Fazzari, P.; Gilestro, G.F.; Barberis, D.; Giordano, S.; Comoglio, P.M.; Tamagnone, L. Plexin-B3 is a functional receptor for semaphorin 5A. EMBO Rep. 2004, 5, 710–714. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lee, A.Y. Semaphorin 5A and plexin-B3 inhibit human glioma cell motility through RhoGDIalpha-mediated inactivation of Rac1 GTPase. J. Biol. Chem. 2010, 285, 32436–32445. [Google Scholar] [CrossRef]
- Li, X.; Law, J.W.; Lee, A.Y. Semaphorin 5A and plexin-B3 regulate human glioma cell motility and morphology through Rac1 and the actin cytoskeleton. Oncogene 2012, 31, 595–610. [Google Scholar] [CrossRef]
- Zhao, J.; Tang, H.; Zhao, H.; Che, W.; Zhang, L.; Liang, P. SEMA6A is a prognostic biomarker in glioblastoma. Tumour Biol. 2015, 36, 8333–8340. [Google Scholar] [CrossRef]
- Formolo, C.A.; Williams, R.; Gordish-Dressman, H.; MacDonald, T.J.; Lee, N.H.; Hathout, Y. Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 2011, 10, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Gould Rothberg, B.E.; Rimm, D.; Scott, G. The semaphorin 7A receptor Plexin C1 is lost during melanoma metastasis. Am. J. Dermatopathol. 2009, 31, 177–181. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828, Erratum in: Nature 2011, 477, 238. Nature 2011, 469, 432. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Machein, M.; de Miguel, L.S. Angiogenesis in gliomas. Recent Results Cancer Res. 2009, 171, 193–215. [Google Scholar] [CrossRef]
- Sica, G.; Lama, G.; Anile, C.; Geloso, M.C.; La Torre, G.; De Bonis, P.; Maira, G.; Lauriola, L.; Jhanwar-Uniyal, M.; Mangiola, A. Assessment of angiogenesis by CD105 and nestin expression in peritumor tissue of glioblastoma. Int. J. Oncol. 2011, 38, 41–49. [Google Scholar] [CrossRef]
- Fan, Y.; Potdar, A.A.; Gong, Y.; Eswarappa, S.M.; Donnola, S.; Lathia, J.D.; Hambardzumyan, D.; Rich, J.N.; Fox, P.L. Profilin-1 phosphorylation directs angiocrine expression and glioblastoma progression through HIF-1α accumulation. Nat. Cell Biol. 2014, 16, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessio, A.; Proietti, G.; Lama, G.; Biamonte, F.; Lauriola, L.; Moscato, U.; Vescovi, A.; Mangiola, A.; Angelucci, C.; Sica, G. Analysis of angiogenesis related factors in glioblastoma, peritumoral tissue and their derived cancer stem cells. Oncotarget 2016, 7, 78541–78556. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Rizzolio, S.; Tamagnone, L. Multifaceted role of neuropilins in cancer. Curr. Med. Chem. 2011, 18, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Taylor, G.I.; Minichiello, J.; Farlie, P.; Cichowitz, A.; Watson, N.; Klagsbrun, M.; Mamluk, R.; Newgreen, D.F. Neurovascular congruence results from a shared patterning mechanism that utilizes Semaphorin3A and Neuropilin-1. Dev. Biol. 2003, 255, 77–98. [Google Scholar] [CrossRef] [Green Version]
- Maione, F.; Molla, F.; Meda, C.; Latini, R.; Zentilin, L.; Giacca, M.; Seano, G.; Serini, G.; Bussolino, F.; Giraudo, E. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J. Clin. Invest. 2009, 119, 3356–3372. [Google Scholar] [CrossRef] [Green Version]
- Barresi, V.; Tuccari, G. Increased ratio of vascular endothelial growth factor to semaphorin3A is a negative prognostic factor in human meningiomas. Neuropathology 2010, 30, 537–546. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, A.; Fu, W.; Lv, Z.; Zhang, Z. Significance of semaphorin-3A and MMP-14 protein expression in non-small cell lung cancer. Oncol. Lett. 2014, 7, 1395–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttmann-Raviv, N.; Shraga-Heled, N.; Varshavsky, A.; Guimaraes-Sternberg, C.; Kessler, O.; Neufeld, G. Semaphorin-3A and semaphorin-3F work together to repel endothelial cells and to inhibit their survival by induction of apoptosis. J. Biol. Chem. 2007, 282, 26294–26305. [Google Scholar] [CrossRef] [PubMed]
- Bassi, D.E.; Fu, J.; Lopezde Cicco, R.; Klein-Szanto, A.J. Proprotein convertases: “master switches” in the regulation of tumor growth and progression. Mol.Carcinog. 2005, 44, 151–161. [Google Scholar] [CrossRef]
- Varshavsky, A.; Kessler, O.; Abramovitch, S.; Kigel, B.; Zaffryar, S.; Akiri, G.; Neufeld, G. Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res. 2008, 68, 6922–6931. [Google Scholar] [CrossRef] [PubMed]
- Karayan-Tapon, L.; Wager, M.; Guilhot, J.; Levillain, P.; Marquant, C.; Clarhaut, J.; Potiron, V.; Roche, J. Semaphorin, neuropilin and VEGF expression in glial tumours: SEMA3G, a prognostic marker? Br. J. Cancer 2008, 99, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Treps, L.; Edmond, S.; Harford-Wright, E.; Galan-Moya, E.M.; Schmitt, A.; Azzi, S.; Citerne, A.; Bidère, N.; Ricard, D.; Gavard, J. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 2016, 35, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Caunt, M.; Mak, J.; Liang, W.C.; Stawicki, S.; Pan, Q.; Tong, R.K.; Kowalski, J.; Ho, C.; Reslan, H.B.; Ross, J.; et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. 2008, 13, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef]
- Miyauchi, J.T.; Tsirka, S.E. Advances in immunotherapeutic research for glioma therapy. J. Neurol. 2018, 265, 741–756. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [CrossRef] [PubMed]
- Kushchayev, S.V.; Kushchayeva, Y.S.; Wiener, P.C.; Scheck, A.C.; Badie, B.; Preul, M.C. Monocyte-derived cells of the brain and malignant gliomas: The double face of Janus. World Neurosurg. 2014, 82, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, R.; Supler, M.L.; Toyka, K.V.; Streit, W.J. In situ detection of transforming growth factor-beta mRNA in experimental rat glioma and reactive glial cells. Neurosci. Lett. 1994, 166, 161–164. [Google Scholar] [CrossRef]
- Kumanogoh, A.; Kikutani, H. The CD100-CD72 interaction: A novel mechanism of immune regulation. Trends Immunol. 2001, 22, 670–676. [Google Scholar] [CrossRef]
- Suzuki, K.; Kumanogoh, A.; Kikutani, H. Semaphorins and their receptors in immune cell interactions. Nat. Immunol. 2008, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.D.; Park-Min, K.H.; Ivashkiv, L.B. Expression and function of semaphorin 3A and its receptors in human monocyte-derived macrophages. Hum. Immunol. 2009, 70, 211–217. [Google Scholar] [CrossRef]
- Catalano, A.; Caprari, P.; Moretti, S.; Faronato, M.; Tamagnone, L.; Procopio, A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood 2006, 107, 3321–3329. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Suzuki, K.; Okuno, T.; Ogata, T.; Takegahara, N.; Takamatsu, H.; Mizui, M.; Taniguchi, M.; Chédotal, A.; Suto, F.; et al. Plexin-A4 negatively regulates T lymphocyte responses. Int. Immunol. 2008, 20, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013, 24, 695–709. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Zhuang, R.; Gao, Z.; Liu, J.; Li, J.; Yang, A.; Cheng, G.; Jin, B. Plexin-B1: A potential diagnostic biomarker for glioma and a future target for glioma immunotherapy. J. Neuroimmunol. 2012, 252, 113–117. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2018, 33, 152. [Google Scholar] [CrossRef]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- McFarland, B.C.; Marks, M.P.; Rowse, A.L.; Fehling, S.C.; Gerigk, M.; Qin, H.; Benveniste, E.N. Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget 2014, 7, 20621–20635. [Google Scholar] [CrossRef]
- Caponegro, M.D.; Moffitt, R.A.; Tsirka, S.E. Expression of neuropilin-1 is linked to glioma associated microglia and macrophages and correlates with unfavorable prognosis in high grade gliomas. Oncotarget 2018, 9, 35655–35665. [Google Scholar] [CrossRef]
- Miyauchi, J.T.; Caponegro, M.D.; Chen, D.; Choi, M.K.; Li, M.; Tsirka, S.E. Deletion of Neuropilin 1 from Microglia or Bone Marrow-Derived Macrophages Slows Glioma Progression. Cancer Res. 2018, 78, 685–694. [Google Scholar] [CrossRef]
- Evans, E.E.; Jonason, A.S., Jr.; Bussler, H.; Torno, S.; Veeraraghavan, J.; Reilly, C.; Doherty, M.A.; Seils, J.; Winter, L.A.; Mallow, C.; et al. Antibody Blockade of Semaphorin 4D Promotes Immune Infiltration into Tumor and Enhances Response to Other Immunomodulatory Therapies. Cancer Immunol. Res. 2015, 3, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Weiss, G.J.; Leonard, J.E.; Rasco, D.W.; Sachdev, J.C.; Fisher, T.L.; Winter, L.A.; Reilly, C.; Parker, R.B.; Mutz, D.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of a Humanized Anti-Semaphorin 4D Antibody, in a First-In-Human Study of Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 827–836. [Google Scholar] [CrossRef]
- Kesari, S. Understanding glioblastoma tumor biology: The potential to improve current diagnosis and treatments. Semin. Oncol. 2011, 4, S2–S10. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Piperi, C. Emerging role of plexins signaling in glioma progression and therapy. Cancer Lett. 2018, 414, 81–87. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Semaphorin | Pro-Tumorigenic | Anti-Tumorigenic |
|---|---|---|
| Sema3A | Increased growth of GBM patient-derived cells (PDCs), in vitro and in vivo [28,29]. NRP1 mediated induction of human A172 and U87MG cell migration via Rac1 activation [30]. Increased migration of PDCs and U87MG cells [27]. Induced recruitment of TAMs [28]. | Reduced proliferation and colony formation of U87MG cells in vitro and inhibition of tumor development in vivo [26]. Inhibition of U87MG cell motility mediated by RhoA inactivation [38]. Inhibition of angiogenesis in U87MG tumors in vivo [26]. |
| Sema3B | _ | Inhibition of U87MG tumor development in vivo [26]. Reduction of U87MG and U251MG cell proliferation, migration and invasiveness [31]. |
| Sema3C | Increased survival and self-renewal of GBM stem cells (GSCs) obtained from tumor specimens via Plexin-A2/D1-mediated Rac1 signaling activation [33]. | _ |
| Sema3D | _ | Inhibition of U87MG tumor development and angiogenesis in vivo [26]. |
| Sema3E | _ | Inhibition of U87MG tumor development and angiogenesis in vivo [26]. |
| Sema3F | _ | Reduced proliferation and colony formation of U87MG cells in vitro [26], and inhibition of tumor development and angiogenesis in vivo [26,32]. NRP2/Plexin-A1 signaling-mediated inhibition of U87MG cell motility via RhoA inactivation [38,39,40,41]. Hypoxia-induced repression of NRP2 expression in GBM U87MG enhanced Sema3F-mediated endothelial cell migration and sprouting [41]. |
| Sema3G | Reduction of U251MG cell migration and invasiveness mediated by the inhibition of metalloproteinase-2 expression [43]. | |
| Sema4C | Increased U87MG and LN229 cell migration in vitro through RhoA and Rac1 activation [44]. Increased tumor growth, invasion, and angiogenesis, and tumor cell perivascular spread via Plexin-B2 signaling in vivo [44]. | _ |
| Sema4D | Increased U87MG and U251 cell viability, migration, and invasiveness via Plexin-B1 signaling [34]. Increased angiogenesis in U87MG xenograft tumors [34]. | Plexin-B1 mediated promotion of natural killer (NK)-cell-mediated killing of tumor cells [82]. |
| Sema5A | _ | Reduction of rat C6 and human U87MG cell migration and invasion via Plexin-B3 mediated Rac1 inactivation [46,47]. |
| Sema6A | _ | Inhibition of U87MG and U251 cell migration and invasion in vitro, probably mediated by Sema6A-induced suppression of ERK1/2 and FAK activation [48]. |
| Sema6B | Plexin-A4-mediated stimulation of U87MG cell proliferation in vitro and tumorigenesis in nude mice [37]. | _ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelucci, C.; Lama, G.; Sica, G. Multifaceted Functional Role of Semaphorins in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2144. https://doi.org/10.3390/ijms20092144
Angelucci C, Lama G, Sica G. Multifaceted Functional Role of Semaphorins in Glioblastoma. International Journal of Molecular Sciences. 2019; 20(9):2144. https://doi.org/10.3390/ijms20092144
Chicago/Turabian StyleAngelucci, Cristiana, Gina Lama, and Gigliola Sica. 2019. "Multifaceted Functional Role of Semaphorins in Glioblastoma" International Journal of Molecular Sciences 20, no. 9: 2144. https://doi.org/10.3390/ijms20092144
APA StyleAngelucci, C., Lama, G., & Sica, G. (2019). Multifaceted Functional Role of Semaphorins in Glioblastoma. International Journal of Molecular Sciences, 20(9), 2144. https://doi.org/10.3390/ijms20092144