Transcriptome Analysis of Apple Leaves in Response to Powdery Mildew (Podosphaera leucotricha) Infection


 and
and
Abstract
:1. Introduction
2. Results
2.1. Statistical Analysis of RNA-seq Results from Different Time Points after PM Inoculation
2.2. DEG Profiles in Response to PM Infection
2.3. Pathway Enrichment Analysis at Different Infection Stages
2.4. DEGs Involved in Phytohormone Signaling
2.5. DEGs Involved in Plant-fungal interaction
2.6. TFs Related to PM Responses
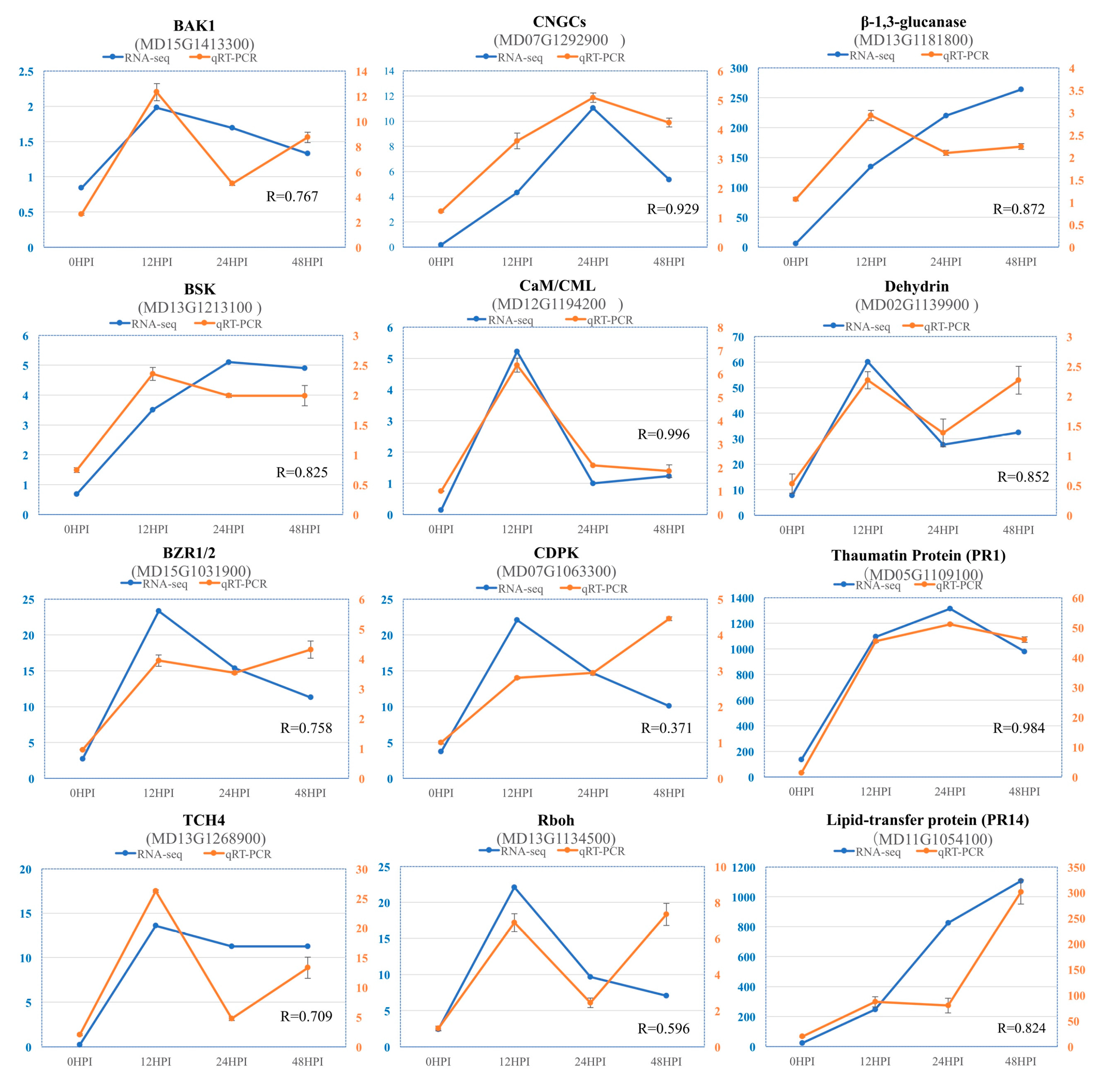
2.7. Validation of RNA-Seq Data by Quantitative Real Time (qRT)-PCR
2.8. Changes in Physiological Characteristics of Apple Leaves after Infection with PM
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Fungal Collection and Inoculation
4.2. RNA Quantification and Qualification
4.3. Library Preparation
4.4. RNA-Seq DataAnalysis
4.5. Differential Expression Analysis
4.6. GO and KEGG Pathway Enrichment Analysis
4.7. qRT-PCR Validation and Analysis
4.8. Effects of PM on Physiological Indexes
4.9. Effects of PM on the Accumulation of Defense-Related Enzyme
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| PM | Powdery mildew |
| DEG | Differentially expressed gene |
| HPI | Hours post inoculation |
| DPI | Days post inoculation |
| PCR | Polymerase chain reaction |
| qRT-PCR | Quantitative real time PCR |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | KEGG Orthology |
| ZF-TFs | Zinc finger-transcription factors |
| PR5 | Thaumatin protein |
| PR10 | Ribonucleases |
| PR14 | Lipid-transfer protein |
| CYPs | Cytochrome P450 genes |
| ET | Ethylene |
| SA | Salicylic acid |
| JA | Jasmonic acid |
| GA | Gibberellin |
| AUX | Auxin |
| BR | brassionosteroid |
| NBT | Nitro blue tetrazolium |
| DAB | Diaminobenzidine |
| Pro | Proline |
| MDA | Malondialdehyde |
| POD | Peroxidase |
| SOD | Superoxide dismutase |
| CAT | Catalase |
| β-1,3-GA | β-1,3-glucanase |
| CHI | Chitinase |
| PAL | Phenylalanine ammonia-lyase |
| ROS | Reactive oxygen species |
References
- Halimie, M.A.; Mughal, M.S.; Aslam, M.; Mehdi, S.A. Effect of fungicides on powdery mildew of apple Podosphaera leucotricha (Ell. and Ev.) Salm. J. Agric. Res. 1992, 30, 259–263. [Google Scholar]
- Turechek, W.W.; Carroll, J.E.; Rosenberger, D.A. Powdery Mildew of Apple. 2005. Available online: https://ecommons.cornell.edu/handle/1813/43120 (accessed on 10 May 2019).
- Szentiványi, O.; Kiss, L. Overwintering of ampelomyces mycoparasites on apple trees and other plants infected with powdery mildews. Plant Pathol. 2010, 52, 737–746. [Google Scholar] [CrossRef]
- Pessina, S.; Angeli, D.; Martens, S.; Visser, R.G.; Bai, Y.; Salamini, F.; Velasco, R.; Schouten, H.J.; Malnoy, M. The knock-down of the expression of MdMLO19 reduces susceptibility to powdery mildew (Podosphaera leucotricha) in apple (Malus domestica). Plant Biotechnol. J. 2016, 14, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97. [Google Scholar] [CrossRef] [PubMed]
- Úrbez-Torres, J.R.; Leavitt, G.M.; Guerrero, J.C.; Guevara, J.; Gubler, W.D. Identification and pathogenicity of lasiodiplodia theobromae and diplodia seriata, the causal agents of bot canker disease of grapevines in Mexico. Plant Dis. 2008, 92, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zheng, X.; Apaliya, M.T.; Yang, H.; Zhang, H. Transcriptome characterization and expression profile of defense-related genes in pear induced by Meyerozyma guilliermondii. Postharv. Biol. Technol. 2018, 141, 63–70. [Google Scholar] [CrossRef]
- Noam, A.; Gilgi, F.; Dana, M.; Dov, P.; Robert, F. Simultaneous transcriptome analysis of colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies. New Phytol. 2015, 205, 801–815. [Google Scholar]
- Michele, G.; Cesare, G.; Broggini, G.A.L. RNA-Seq analysis reveals candidate genes for ontogenic resistance in malus-venturia pathosystem. PLoS ONE 2013, 8, e78457. [Google Scholar]
- Zhu, L.; Ni, W.; Liu, S.; Cai, B.; Xing, H.; Wang, S. Transcriptomics Analysis of apple leaves in response to alternaria alternata apple pathotype infection. Front. Plant Sci. 2017, 8, 22. [Google Scholar] [CrossRef]
- Jianhua, X.; Miaomiao, L.; Peng, J.; Hongxia, T.; Ningning, W.; Fengwang, M.; Junke, Z. Dynamic transcription profiles of “Qinguan” apple (malus × domestica) leaves in response to Marssonina coronaria inoculation. Front. Plant Sci. 2015, 6, 842. [Google Scholar]
- Yin, Z.; Ke, X.; Kang, Z.; Huang, L. Apple resistance responses against Valsa mali revealed by transcriptomics analyses. Physiol. Mol. Plant Pathol. 2016, 93, 85–92. [Google Scholar] [CrossRef]
- Shin, S.; Zheng, P.; Fazio, G.; Mazzola, M.; Main, D.; Zhu, Y. Transcriptome changes specifically associated with apple (malus domestica) root defense response during Pythium ultimum infection. Physiol. Mol. Plant Pathol. 2016, 94, 16–26. [Google Scholar] [CrossRef]
- Urbanietz, A.; Dunemann, F. Isolation, identification and molecular characterization of physiological races of apple powdery mildew (Podosphaera leucotricha). Plant Pathol. 2010, 54, 125–133. [Google Scholar] [CrossRef]
- Vivancos, J.; Labbã, C.; Menzies, J.G.; Bã Langer, R.R. Silicon-mediated resistance of Arabidopsis against powdery mildew involves mechanisms other than the salicylic acid (SA)-dependent defence pathway. Mol. Plant Pathol. 2015, 16, 572–582. [Google Scholar] [CrossRef]
- Malnoy, M.; Jin, Q.; Borejsza-Wysocka, E.E.; He, S.Y.; Aldwinckle, H.S. Overexpression of the apple MpNPR1 gene confers increased disease resistance in Malus x domestica. Mol. Plant Microbe Interact. 2007, 20, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, V.; Andrey, Z.; Jason, A.; Amit, D.; Alessandro, C.; Ananth, K.; Paolo, F.; Bhatnagar, S.K.; Michela, T.; Dmitry, P. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [Green Version]
- Pieterse, C.M.; Leon-Reyes, A.; Ent van der, S.; van Wees, S.C. Networking by small-molecule hormones in plant immunity. Nat. Chem. Biol. 2009, 5, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidhyasekaran, P. Molecular Manipulation of Transcription Factors, the Master Regulators of PAMP-Triggered Signaling Systems. In Switching on Plant Innate Immunity Signaling Systems; Springer: Berlin/Heidelberg, Germany, 2016; pp. 255–358. [Google Scholar]
- Lamb, C.J.; Brisson, L.; Bradley, D.J.; Kjellbom, P. Stimulus-Dependent Oxidative Cross-Linking of a Proline-Rich Plant Cell Wall Protein: A Novel, Rapid Defense Response and Control Point in Cellular Maturation. J. Cell. Biochem. 1993, 2, 4. [Google Scholar]
- Berger, S.; Sinha, A.K.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactions. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef]
- Lei, M.Y.; Hui-Liang, L.I.; Liu, L.Y.; Peng, S.Q. Sugar sensing and signaling in plants. Acta Botanica Boreali-Occidentalia Sinica 2007, 10, S185–S205. [Google Scholar]
- Meloni, D.A.; Oliva, M.A.; Martinez, C.A.; Cambraia, J. Photosynthesis and activity of superoxide dismutase, peroxidase and glutathione reductase in cotton under salt stress. Environ. Exp. Bot. 2003, 49, 69–76. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Ann. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Juan, Y.; Su-Su, Y.; Luan-Luan, J.; Xiu-Juan, Y.; Tzi Bun, N.; Zu-Jian, W. Plant antifungal proteins and their applications in agriculture. Appl. Microbiol. Biotechnol. 2015, 99, 4961–4981. [Google Scholar]
- Riet, K.B.; Ndlovu, N.; Piater, L.A.; Dubery, I.A. Simultaneous analysis of defense-related phytohormones in Arabidopsis thaliana responding to fungal infection. Appl. Plant Sci. 2016, 4, 1600013. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.; Fujioka, S.; Dilkes, B.P.; Choe, S. Brassinosteroids regulate plant growth through distinct signaling pathways in Selaginella and Arabidopsis. PLoS ONE 2013, 8, e81938. [Google Scholar] [CrossRef]
- David, D.V.; Godelieve, G.; Monica, H.F. Hormone defense networking in rice: Tales from a different world. Trends Plant Sci. 2013, 18, 555–565. [Google Scholar]
- Kunkel, B.N.; Brooks, D.M. Cross talk between signaling pathways in pathogen defense. Curr. Opin. Plant Biol. 2002, 5, 325–331. [Google Scholar] [CrossRef]
- van loon, L.C. Pathogenesis-Related Proteins, Plant Molecular Biology 1985, 4, 209–243.
- Ignatius, S.M.J.; Chopra, R.K.; Muthukrishnan, S. Effects of fungal infection and wounding on the expression of chitinases and β-1,3 glucanases in near-isogenic lines of barley. Physiol. Plant. 2010, 90, 584–592. [Google Scholar] [CrossRef]
- Fujimori, N.; Enoki, S.; Suzuki, A.; Naznin, H.A.; Shimizu, M.; Suzuki, S. Grape apoplasmic β-1,3-glucanase confers fungal disease resistance in Arabidopsis. Sci. Horticult. 2016, 200, 105–110. [Google Scholar] [CrossRef]
- Ma, W.; Berkowitz, G.A. The grateful dead: calcium and cell death in plant innate immunity. Cell. Microbiol. 2010, 9, 2571–2585. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, C.; Yong, D.; Li, G.; Dong, X.; Li, B. Induction of resistance mediated by an attenuated strain of valsa mali var. mali using pathogen-apple callus interaction system. Sci. World J. 2014. [Google Scholar] [CrossRef]
- Reuber, T.L.; Plotnikova, J.M.; Dewdney, J.; Rogers, E.E.; Wood, W.; Ausubel, F.M. Correlation of defense gene induction defects with powdery mildew susceptibility in Arabidopsis enhanced disease susceptibility mutants. Plant J. 2010, 16, 473–485. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef]
- Maldonado-Bonilla, L.D.; Lennart, E.L.; Selma, G.Z.; Naheed, T.; Nicole, B.; Dierk, S.; Justin, L. The arabidopsis tandem zinc finger 9 protein binds RNA and mediates pathogen-associated molecular pattern-triggered immune responses. Plant Cell Physiol. 2014, 55, 412–425. [Google Scholar] [CrossRef]
- Gamboameléndez, H.; Huerta, A.I.; Judelson, H.S. bZIP transcription factors in the oomycete phytophthora infestans with novel DNA-binding domains are involved in defense against oxidative stress. Eukaryot. Cell 2013, 12, 1403–1412. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Li, D.; Huang, L.; Hong, Y.; Ding, X.S.; Nelson, R.S.; Zhou, X.; Song, F. Functions of rice NAC transcriptional factors, ONAC122 and ONAC131, in defense responses against Magnaporthe grisea. Plant Mol. Biol. 2013, 81, 41–56. [Google Scholar] [CrossRef]
- Girardi, C.L.; Rombaldi, C.V.; Cero, J.D.; Nobile, P.M.; Laurens, F.; Bouzayen, M.; Quecini, V. Genome-wide analysis of the AP2/ERF superfamily in apple and transcriptional evidence of ERF involvement in scab pathogenesis. Sci. Horticult. 2013, 151, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.A.; Achnine, L.; Kota, P.; Liu, C.J.; Reddy, M.S.; Wang, L. The phenylpropanoid pathway and plant defence-a genomics perspective. Mol. Plant Pathol. 2010, 3, 371–390. [Google Scholar] [CrossRef]
- Corina, W.; Ulrich, H.; Thomas, H.; Christoph, H.; Sebastian, P.; Constanze, K.N.; Wilfried, S.; Ruth, E.; Ralph, H. CYP83A1 is required for metabolic compatibility of Arabidopsis with the adapted powdery mildew fungus Erysiphe cruciferarum. New Phytol. 2014, 202, 1310–1319. [Google Scholar]
- Liu, Y.; Shi, C.; Mu, X.; Chao, L.; Ke, S.; Zhu, W.; Yang, Q. Cloning and expression of a wild eggplant cytochrome P450 gene, StoCYP77A2, involved in plant resistance to Verticillium dahliae. Plant Biotechnol. Rep. 2015, 9, 167–177. [Google Scholar]
- Hwang, I.S.; Hwang, B.K. Role of the pepper cytochrome P450 gene CaCYP450A in defense responses against microbial pathogens. Planta 2010, 232, 1409. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Finn, R.D.; Alex, B.; Jody, C.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. Nucl. Acids Res. 2014, 42, 222–230. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucl. Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucl. Acids Res. 2000, 28, 45. [Google Scholar] [CrossRef]
- Minoru, K.; Michihiro, A.; Susumu, G.; Masahiro, H.; Mika, H.; Masumi, I.; Toshiaki, K.; Shuichi, K.; Shujiro, O.; Toshiaki, T. KEGG for linking genomes to life and the environment. Nucl. Acids Res. 2008, 36, D480–D484. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Hardcastle, T.J.; Kelly, K.A. baySeq: Empirical bayesian methods for identifying differential expression in sequence count data. BMC Bioinf. 2010, 11, 422. [Google Scholar] [CrossRef]
- Genovese, C.R.; Lazar, N.A.; Thomas, N. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 2002, 15, 870–878. [Google Scholar] [CrossRef]
- Mao, X.; Tao, C.J.G.O.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 −ΔΔ CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Zude-Sasse, M.; Truppel, I.; Herold, B. An approach to non-destructive apple fruit chlorophyll determination. Postharv. Biol. Technol. 2002, 25, 123–133. [Google Scholar] [CrossRef]
- Lu, W.; Yan, L.; Hong, S.; Li, X. Comparison of carbohydrate content variation between wild species and cultivars of rehmannia glutinosa. Chin. Agric. Sci. Bullet. 2013, 29, 147–153. [Google Scholar]
- Qingdian, H.; Xiaojun, H.; Kunyan, H. Effects in Activities of Defense Enzymes and Contents of MDA in Wheat Leaf Infected by Powdery Mildew. Mol. Plant Breed. 2016. [Google Scholar]
- Yong, D.; Wang, C.; Li, G.; Li, B. Control efficiency of endophytic actinomycetes A-1 against apple fruit ring rot and its influence on the activity of defense-related enzymes. Acta Phytophylacica Sinica 2014, 41, 335–341. [Google Scholar]
- Lu, L.; Lu, H.; Wu, C.; Fang, W.; Chen, Y.; Ye, C.; Shi, Y.; Yu, T.; Zheng, X. Rhodosporidium paludigenum induces resistance and defense-related responses against Penicillium digitatum in citrus fruit. Postharv. Biol. Technol. 2013, 85, 196–202. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Time Points | Total Reads | Q20 Percentage (%) | Q30 Percentage (%) | Mapped Reads | Mapping Rate (%) |
|---|---|---|---|---|---|---|
| CK01 | 0 hpi | 60,137,610 | 97.4 | 93.65 | 48,293,530 | 80.31 |
| CK02 | 0 hpi | 61,254,152 | 97.5 | 93.83 | 49,772,328 | 81.26 |
| CK03 | 0 hpi | 58,546,454 | 97.46 | 93.77 | 47,250,937 | 80.71 |
| CK121 | 12 hpi | 48,161,690 | 97.55 | 94.05 | 37,977,254 | 78.85 |
| CK122 | 12 hpi | 60,404,448 | 96.95 | 92.80 | 47,385,808 | 78.85 |
| CK123 | 12 hpi | 62,016,814 | 97.29 | 93.41 | 49,706,083 | 80.15 |
| CK241 | 24 hpi | 54,165,314 | 96.78 | 92.46 | 42,586,645 | 78.62 |
| CK242 | 24 hpi | 64,515,854 | 96.86 | 92.67 | 51,035,900 | 79.11 |
| CK243 | 24 hpi | 70,503,200 | 96.76 | 92.47 | 55,360,754 | 78.52 |
| CK481 | 48 hpi | 64,182,946 | 97.06 | 92.98 | 51,398,939 | 80.08 |
| CK482 | 48 hpi | 56,164,490 | 96.91 | 92.69 | 44,903,336 | 79.95 |
| CK483 | 48 hpi | 56,864,248 | 96.76 | 92.42 | 44,919,501 | 78.99 |
| T01 | 0 hpi | 68,815,498 | 97.41 | 93.67 | 56,225,369 | 78.99 |
| T02 | 0 hpi | 60,247,576 | 97.12 | 93.17 | 48,129,005 | 78.99 |
| T03 | 0 hpi | 60,534,844 | 97.13 | 93.16 | 48,533,584 | 78.99 |
| T121 | 12 hpi | 55,781,200 | 96.74 | 92.89 | 44,030,266 | 78.99 |
| T122 | 12 hpi | 68,568,070 | 97.03 | 92.94 | 54,673,257 | 78.99 |
| T123 | 12 hpi | 53,809,766 | 97.21 | 93.29 | 43,094,498 | 80.09 |
| T241 | 24 hpi | 66,547,810 | 96.74 | 92.47 | 52,184,112 | 78.42 |
| T242 | 24 hpi | 55,181,416 | 97.03 | 92.96 | 43,627,644 | 79.06 |
| T243 | 24 hpi | 43,835,550 | 96.86 | 92.63 | 34,502,703 | 78.71 |
| T481 | 48 hpi | 56,609,920 | 96.89 | 92.69 | 45,122,462 | 79.71 |
| T482 | 48 hpi | 52,151,048 | 96.9 | 92.77 | 40,989,977 | 78.60 |
| T483 | 48 hpi | 71,638,240 | 96.81 | 92.55 | 56,853,084 | 79.36 |
| Average | 59,609,923 | 97.05 | 93.02 | 47,439,874 | 79.35 |
| Pathway Name | Number of Genes with Pathway Annotation | ||||
|---|---|---|---|---|---|
| No. | Pathway ID | G12 | G24 | G48 | |
| Plant hormone signal transduction | P1 | KO 04075 | 15 | 10 | 5 |
| Plant-pathogen interaction | P2 | KO 04626 | 11 | 8 | 1 |
| Phenylpropanoid biosynthesis | P3 | KO 00940 | 10 | 8 | 5 |
| Cyanoamino acid metabolism | P4 | KO 00460 | 7 | 8 | 3 |
| ABC transporters | P5 | KO 02010 | 6 | 7 | 1 |
| Protein processing in endoplasmic reticulum | P6 | KO 04141 | 5 | 7 | 1 |
| Starch and sucrose metabolism | P7 | KO 00500 | 4 | 5 | 3 |
| Cysteine and methionine metabolism | P8 | KO 00270 | 4 | 4 | 7 |
| Phagosome | P9 | KO 04145 | 4 | 3 | 1 |
| Phenylalanine metabolism | P10 | KO 00360 | 3 | 3 | 2 |
| Endocytosis | P11 | KO 04144 | 3 | 3 | 1 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | P12 | KO 00945 | 3 | 3 | 1 |
| alpha-Linolenic acid metabolism | P13 | KO 00592 | 3 | 3 | 0 |
| Ubiquitin mediated proteolysis | P14 | KO 04120 | 3 | 3 | 0 |
| DNA replication | P15 | KO 03030 | 3 | 3 | 0 |
| Lysine degradation | P16 | KO 00310 | 3 | 3 | 0 |
| Linoleic acid metabolism | P17 | KO 00591 | 3 | 3 | 0 |
| Glutathione metabolism | P18 | KO 00480 | 2 | 3 | 1 |
| Sesquiterpenoid and triterpenoid biosynthesis | P19 | KO 00909 | 2 | 1 | 1 |
| Biosynthesis of amino acids | P20 | KO 01230 | 2 | 0 | 1 |
| Glycine, serine and threonine metabolism | P21 | KO 00260 | 2 | 1 | 1 |
| Arginine and proline metabolism | P22 | KO 00330 | 2 | 1 | 1 |
| Steroid biosynthesis | P23 | KO 00100 | 2 | 1 | 3 |
| Carbon metabolism | P24 | KO 01200 | 2 | 2 | 0 |
| SNARE interactions in vesicular transport | P25 | KO 04130 | 2 | 1 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, X.; Zhang, L.; Feng, S.; Zhao, Z.; Wang, X.; Gao, H. Transcriptome Analysis of Apple Leaves in Response to Powdery Mildew (Podosphaera leucotricha) Infection. Int. J. Mol. Sci. 2019, 20, 2326. https://doi.org/10.3390/ijms20092326
Tian X, Zhang L, Feng S, Zhao Z, Wang X, Gao H. Transcriptome Analysis of Apple Leaves in Response to Powdery Mildew (Podosphaera leucotricha) Infection. International Journal of Molecular Sciences. 2019; 20(9):2326. https://doi.org/10.3390/ijms20092326
Chicago/Turabian StyleTian, Xiaomin, Li Zhang, Shuaishuai Feng, Zhengyang Zhao, Xiping Wang, and Hua Gao. 2019. "Transcriptome Analysis of Apple Leaves in Response to Powdery Mildew (Podosphaera leucotricha) Infection" International Journal of Molecular Sciences 20, no. 9: 2326. https://doi.org/10.3390/ijms20092326
APA StyleTian, X., Zhang, L., Feng, S., Zhao, Z., Wang, X., & Gao, H. (2019). Transcriptome Analysis of Apple Leaves in Response to Powdery Mildew (Podosphaera leucotricha) Infection. International Journal of Molecular Sciences, 20(9), 2326. https://doi.org/10.3390/ijms20092326