Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension

Abstract
:1. Introduction
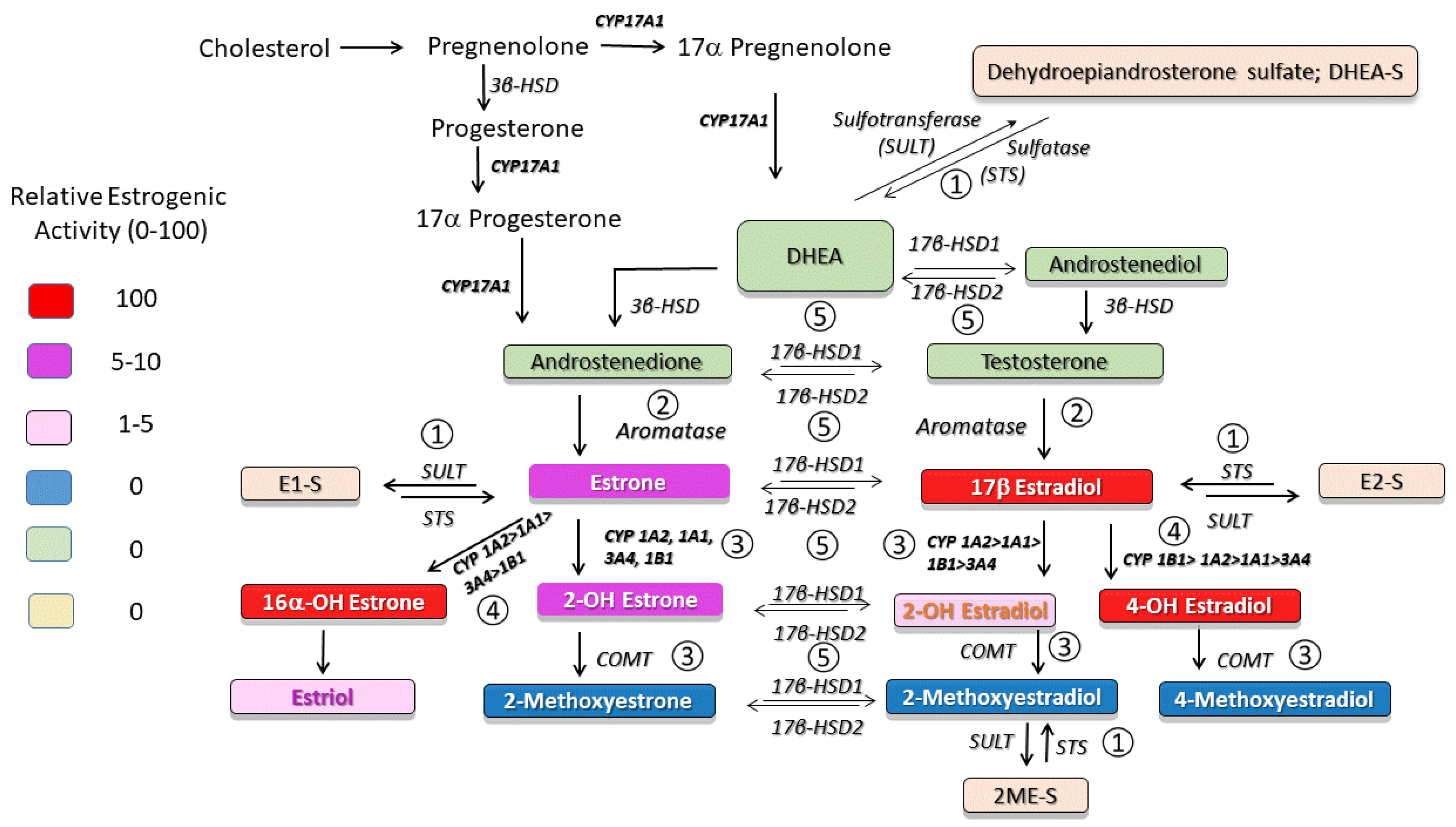
2. Sulfotransferase (SULT)–Sulfatase (STS) Pathway
2.1. SULT–STS Pathway in PAH
2.2. Future Directions and Clinical Implications
3. Aromatase Pathway
3.1. Aromatase in PAH
3.2. Aromatase Inhibition and RV Function in PAH
3.3. Future Directions and Clinical Implications
4. 2-Hydroxylation/2–Methylation Pathway
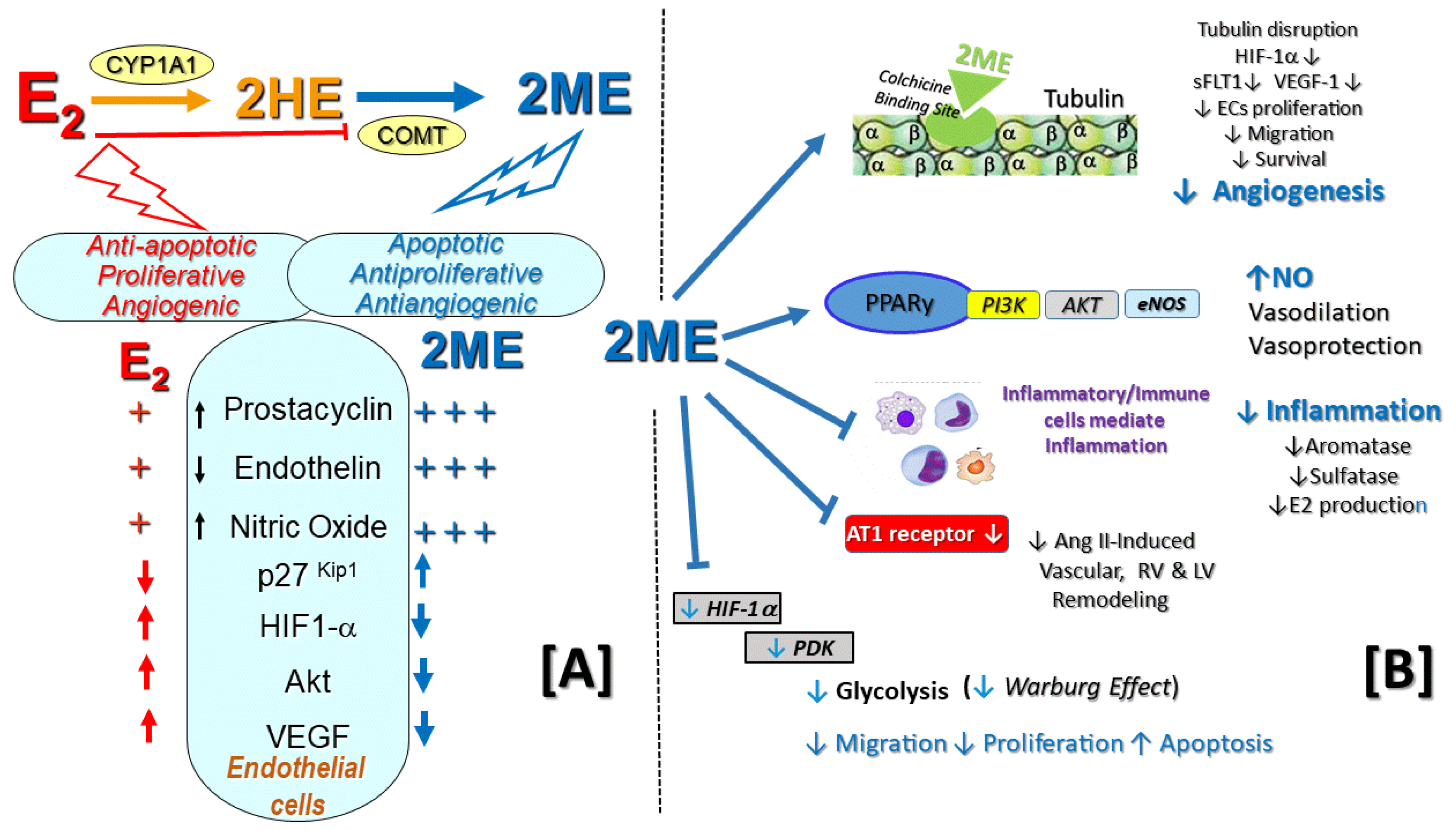
4.1. 2-Hydroxylation/2-Methylation Pathway in PAH
4.2. Other Effects of the 2-Methylation Pathway Relevant to PAH
4.3. Future Directions and Clinical Implications
5. 4- and 16α-Hydroxylation Pathways
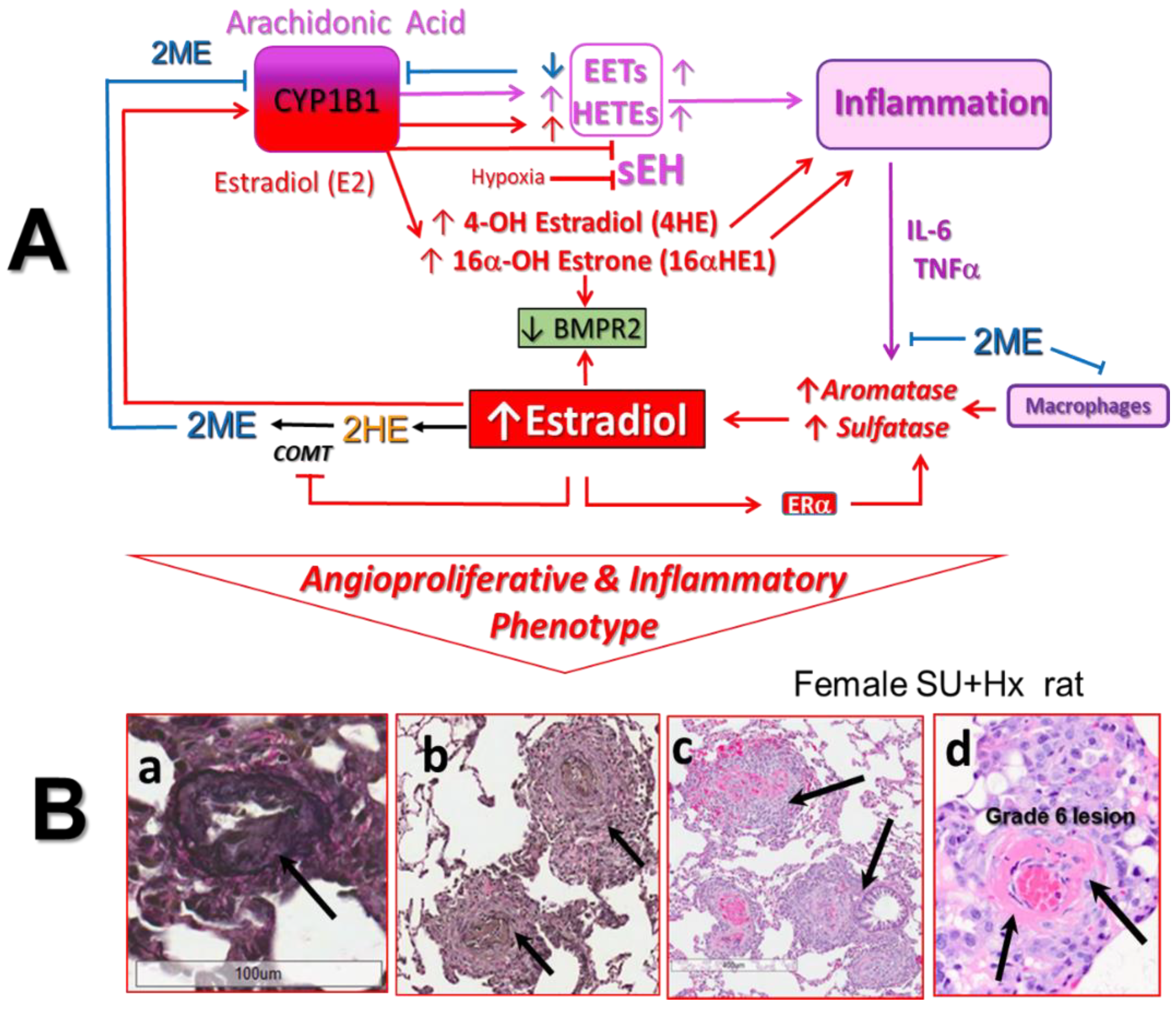
5.1. Role of CYP1B1 in PAH
5.2. Divergent Effects on E2 and 2ME on CYP1B1 Activity
5.3. Role of CYP1B1 and Estrogens in Arachidonic Acid Metabolism
5.4. Future Directions and Clinical Implications
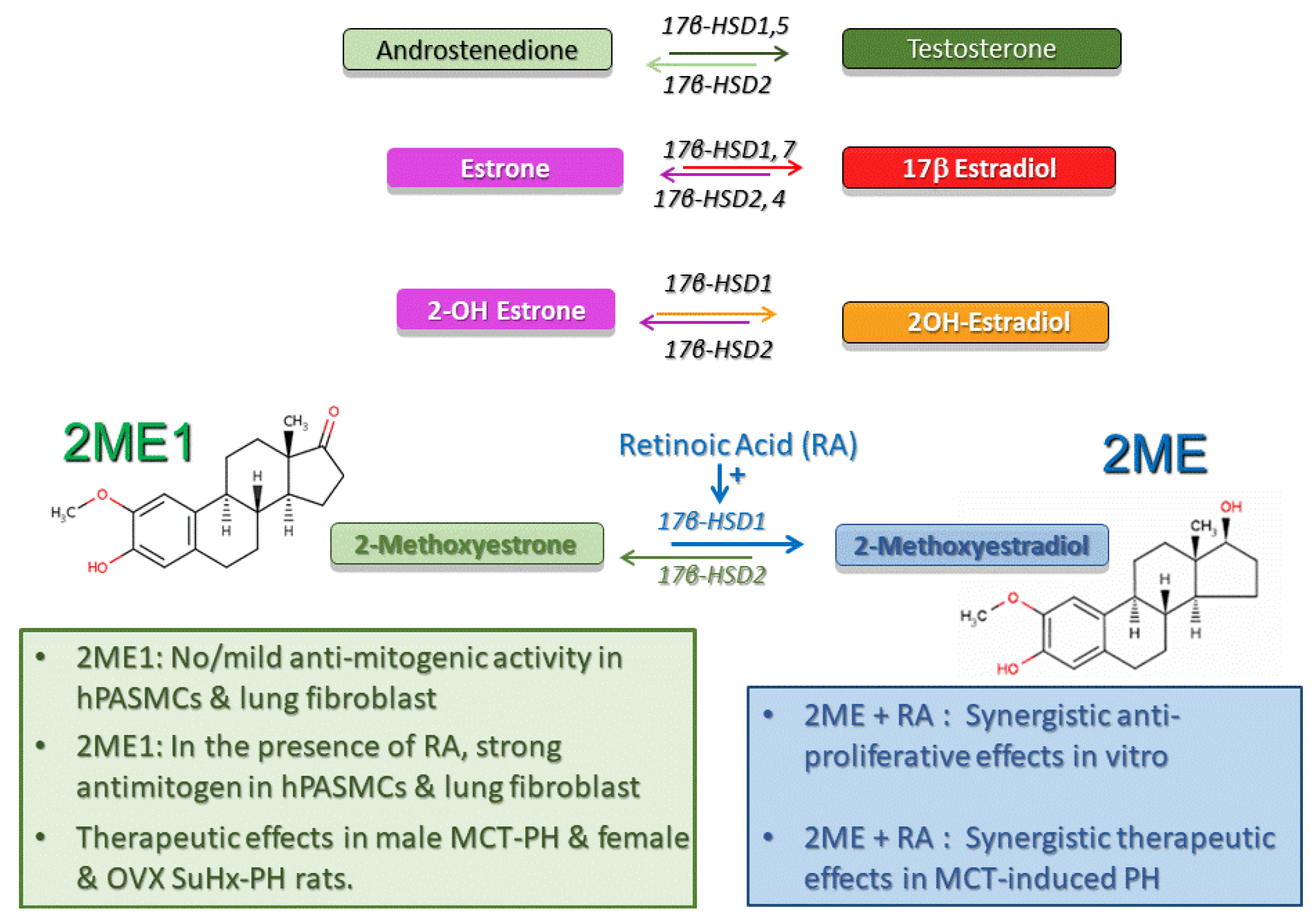
6. 17β-Hydroxysteroid Dehydrogenase Pathway
6.1. 17β-HSD Pathway and 2ME Disposition
6.2. 17β–HSD Pathway in Experimental PH
6.3. Future Directions and Clinical Implications
7. Angiogenesis, Metabolic Reprograming, and Estradiol Metabolism in PAH
7.1. Opposing Effects of E2 and 2ME on Angiogenesis (Key Role of HIF-1α)
7.2. 2ME, HIF1α, and Endothelial-to-Mesenchymal Transition in PAH
7.3. 2ME, HIF-1α, and Metabolic Reprograming in PAH
7.4. Future Directions and Clinical Implications
8. Inflammation, Immunity, and Estradiol Metabolism in PAH
8.1. Anti-Inflammatory and Immunomodulatory Effects of 2ME
8.2. Future Directions and Clinical Implications
9. Concluding Remarks and Future Directions
Funding
Conflicts of Interest
References
- Dresdale, D.T.; Schultz, M.; Michtom, R.J. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am. J. Med. 1951, 11, 686–705. [Google Scholar] [CrossRef] [Green Version]
- Wood, P. Pulmonary hypertension. Br. Med. Bull. 1952, 8, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Kumamaru, H.; Satoh, T.; Miyata, H.; Ogawa, A.; Tanabe, N.; Hatano, M.; Yao, A.; Abe, K.; Tsujino, I.; et al. Effectiveness and Outcome of Pulmonary Arterial Hypertension-Specific Therapy in Japanese Patients With Pulmonary Arterial Hypertension. Circ. J. 2017, 82, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Batton, K.A.; Austin, C.O.; Bruno, K.A.; Burger, C.D.; Shapiro, B.P.; Fairweather, D. Sex differences in pulmonary arterial hypertension: Role of infection and autoimmunity in the pathogenesis of disease. Biol. Sex Differ. 2018, 9, 15. [Google Scholar] [CrossRef]
- Chung, L.; Farber, H.W.; Benza, R.; Miller, D.P.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Nicolls, M.R.; Zamanian, R.T. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. Chest 2014, 146, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880. [Google Scholar] [CrossRef]
- Tofovic, S.P. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J. Cardiovasc. Pharmacol. 2010, 56, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Colvin, K.L.; Yeager, M.E. Animal Models of Pulmonary Hypertension: Matching Disease Mechanisms to Etiology of the Human Disease. J. Pulm. Respir. Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Maarman, G.; Lecour, S.; Butrous, G.; Thienemann, F.; Sliwa, K. A comprehensive review: The evolution of animal models in pulmonary hypertension research; are we there yet? Pulm. Circ. 2013, 3, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Salah, E.M.; Mady, H.H.; Jackson, E.K.; Melhem, M.F. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J. Cardiovasc. Pharmacol. 2005, 46, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Frump, A.L.; Albrecht, M.E.; Fisher, A.J.; Cook, T.G.; Jones, T.J.; Yakubov, B.; Whitson, J.; Fuchs, R.K.; Liu, A.; et al. 17beta-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L375–L388. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Ouyang, P.; Bluemke, D.A.; Tandri, H.; Barr, R.G.; Bagiella, E.; Cappola, A.R.; Bristow, M.R.; Johnson, C.; Kronmal, R.A.; et al. Sex hormones are associated with right ventricular structure and function: The MESA-right ventricle study. Am. J. Respir. Crit. Care Med. 2011, 183, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Frump, A.L.; Goss, K.N.; Vayl, A.; Albrecht, M.; Fisher, A.; Tursunova, R.; Fierst, J.; Whitson, J.; Cucci, A.R.; Brown, M.B.; et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: Effects of endogenous and exogenous sex hormones. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L873–L890. [Google Scholar] [CrossRef]
- Lahm, T.; Douglas, I.S.; Archer, S.L.; Bogaard, H.J.; Chesler, N.C.; Haddad, F.; Hemnes, A.R.; Kawut, S.M.; Kline, J.A.; Kolb, T.M.; et al. Assessment of Right Ventricular Function in the Research Setting: Knowledge Gaps and Pathways Forward. An Official American Thoracic Society Research Statement. Am. J. Respir. Crit. Care Med. 2018, 198, e15–e43. [Google Scholar] [CrossRef] [Green Version]
- Badlam, J.B.; Austin, E.D. Beyond oestrogens: Towards a broader evaluation of the hormone profile in pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Jackson, E.K. Estrogens in Men: Another Layer of Complexity of Estradiol Metabolism in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 193, 1087–1090. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Lahm, T.; West, J.; Tofovic, S.P.; Johansen, A.K.; Maclean, M.R.; Alzoubi, A.; Oka, M. Gender, sex hormones and pulmonary hypertension. Pulm. Circ. 2013, 3, 294–314. [Google Scholar] [CrossRef] [Green Version]
- Tofovic, S.P.; Zhang, X.; Jackson, E.K.; Dacic, S.; Petrusevska, G. 2-Methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul. Pharmacol. 2006, 45, 358–367. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Zhang, X.; Jackson, E.K.; Zhu, H.; Petrusevska, G. 2-methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul. Pharmacol. 2009, 51, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofovic, S.P.; Zhang, X.; Zhu, H.; Jackson, E.K.; Rafikova, O.; Petrusevska, G. 2-Ethoxyestradiol is antimitogenic and attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling. Vascul. Pharmacol. 2008, 48, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Tuder, R.M.; Petrache, I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L7–L26. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Kawut, S.M. Inhibiting oestrogen signalling in pulmonary arterial hypertension: Sex, drugs and research. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, J.W.; Gilligan, L.C.; Idkowiak, J.; Arlt, W.; Foster, P.A. The Regulation of Steroid Action by Sulfation and Desulfation. Endocr. Rev. 2015, 36, 526–563. [Google Scholar] [CrossRef]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Radford, D.J.; Wang, K.; McNelis, J.C.; Taylor, A.E.; Hechenberger, G.; Hofmann, J.; Chahal, H.; Arlt, W.; Lord, J.M. Dehydroepiandrosterone sulfate directly activates protein kinase C-beta to increase human neutrophil superoxide generation. Mol. Endocrinol. 2010, 24, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Strott, C.A. Steroid sulfotransferases. Endocr. Rev. 1996, 17, 670–697. [Google Scholar] [CrossRef]
- Garbacz, W.G.; Jiang, M.; Xie, W. Sex-Dependent Role of Estrogen Sulfotransferase and Steroid Sulfatase in Metabolic Homeostasis. Adv. Exp. Med. Biol. 2017, 1043, 455–469. [Google Scholar] [CrossRef]
- Spink, B.C.; Katz, B.H.; Hussain, M.M.; Pang, S.; Connor, S.P.; Aldous, K.M.; Gierthy, J.F.; Spink, D.C. SULT1A1 catalyzes 2-methoxyestradiol sulfonation in MCF-7 breast cancer cells. Carcinogenesis 2000, 21, 1947–1957. [Google Scholar] [CrossRef] [Green Version]
- Prough, R.A.; Clark, B.J.; Klinge, C.M. Novel mechanisms for DHEA action. J. Mol. Endocrinol. 2016, 56, R139–R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcu, A.; Smith, J.M.; Auchus, R.; Rainey, W.E. Adrenal androgens and androgen precursors-definition, synthesis, regulation and physiologic actions. Compr. Physiol. 2014, 4, 1369–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forney, J.P.; Milewich, L.; Chen, G.T.; Garlock, J.L.; Schwarz, B.E.; Edman, C.D.; MacDonald, P.C. Aromatization of androstenedione to estrone by human adipose tissue in vitro. Correlation with adipose tissue mass, age, and endometrial neoplasia. J. Clin. Endocrinol. Metab. 1981, 53, 192–199. [Google Scholar] [CrossRef]
- Kley, H.K.; Deselaers, T.; Peerenboom, H.; Kruskemper, H.L. Enhanced conversion of androstenedione to estrogens in obese males. J. Clin. Endocrinol. Metab. 1980, 51, 1128–1132. [Google Scholar] [CrossRef]
- Dessouroux, A.; Akwa, Y.; Baulieu, E.E. DHEA decreases HIF-1alpha accumulation under hypoxia in human pulmonary artery cells: Potential role in the treatment of pulmonary arterial hypertension. J. Steroid Biochem. Mol. Biol. 2008, 109, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Dillon, J.S. Dehydroepiandrosterone stimulates nitric oxide release in vascular endothelial cells: Evidence for a cell surface receptor. Steroids 2004, 69, 279–289. [Google Scholar] [CrossRef]
- Bonnet, S.; Dumas-de-La-Roque, E.; Begueret, H.; Marthan, R.; Fayon, M.; Santos, P.D.; Savineau, J.P.; Baulieu, E.E. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2003, 100, 9488–9493. [Google Scholar] [CrossRef] [Green Version]
- Hampl, V.; Bibova, J.; Povysilova, V.; Herget, J. Dehydroepiandrosterone sulphate reduces chronic hypoxic pulmonary hypertension in rats. Eur. Respir. J. 2003, 21, 862–865. [Google Scholar] [CrossRef]
- Oka, M.; Karoor, V.; Homma, N.; Nagaoka, T.; Sakao, E.; Golembeski, S.M.; Limbird, J.; Imamura, M.; Gebb, S.A.; Fagan, K.A.; et al. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc. Res. 2007, 74, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Homma, N.; Nagaoka, T.; Karoor, V.; Imamura, M.; Taraseviciene-Stewart, L.; Walker, L.A.; Fagan, K.A.; McMurtry, I.F.; Oka, M. Involvement of RhoA/Rho kinase signaling in protection against monocrotaline-induced pulmonary hypertension in pneumonectomized rats by dehydroepiandrosterone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L71–L78. [Google Scholar] [CrossRef] [Green Version]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef]
- Sharma, D.; Coridon, H.; Aubry, E.; Houeijeh, A.; Houfflin-Debarge, V.; Besson, R.; Deruelle, P.; Storme, L. Vasodilator effects of dehydroepiandrosterone (DHEA) on fetal pulmonary circulation: An experimental study in pregnant sheep. PLoS ONE 2018, 13, e0198778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Roque, E.D.; Quignard, J.F.; Ducret, T.; Dahan, D.; Courtois, A.; Begueret, H.; Marthan, R.; Savineau, J.P. Beneficial effect of dehydroepiandrosterone on pulmonary hypertension in a rodent model of pulmonary hypertension in infants. Pediatr. Res. 2013, 74, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzoubi, A.; Toba, M.; Abe, K.; O’Neill, K.D.; Rocic, P.; Fagan, K.A.; McMurtry, I.F.; Oka, M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1708–H1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Roque, E.D.; Savineau, J.P.; Metivier, A.C.; Billes, M.A.; Kraemer, J.P.; Doutreleau, S.; Jougon, J.; Marthan, R.; Moore, N.; Fayon, M.; et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): A pilot study. Ann. Endocrinol. 2012, 73, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Baird, G.L.; Barr, R.G.; Bluemke, D.A.; Fritz, J.S.; Hill, N.S.; Klinger, J.R.; Lima, J.A.; Ouyang, P.; Palevsky, H.I.; et al. Higher Estradiol and Lower Dehydroepiandrosterone-Sulfate Levels Are Associated with Pulmonary Arterial Hypertension in Men. Am. J. Respir. Crit. Care Med. 2016, 193, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.L.; Archer-Chicko, C.; Barr, R.G.; Bluemke, D.A.; Foderaro, A.E.; Fritz, J.S.; Hill, N.S.; Kawut, S.M.; Klinger, J.R.; Lima, J.A.C.; et al. Lower DHEA-S levels predict disease and worse outcomes in post-menopausal women with idiopathic, connective tissue disease- and congenital heart disease-associated pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1800467. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Wu, W.H.; Gao, L.; Zheng, Z.Q.; Liu, D.; Mei, H.Y.; Zhang, Z.L.; Jing, Z.C. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, prostacyclin and endothelin-1 pathways. Eur. Respir. J. 2013, 41, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Rafikova, O.; Rafikov, R.; Meadows, M.L.; Kangath, A.; Jonigk, D.; Black, S.M. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm. Circ. 2015, 5, 184–197. [Google Scholar] [CrossRef] [Green Version]
- De la Roque, E.D.; Bellance, N.; Rossignol, R.; Begueret, H.; Billaud, M.; Santos, P.D.; Ducret, T.; Marthan, R.; Dahan, D.; Ramos-Barbon, D.; et al. Dehydroepiandrosterone reverses chronic hypoxia/reoxygenation-induced right ventricular dysfunction in rats. Eur. Respir. J. 2012, 40, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.T.; Xue, J.J.; Wang, Q.; Cheng, S.Y.; Chen, Z.C.; Li, H.Y.; Shan, J.J.; Cheng, K.L.; Zeng, W.J. Dehydroepiandrosterone attenuates pulmonary artery and right ventricular remodeling in a rat model of pulmonary hypertension due to left heart failure. Life Sci. 2019, 219, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Yuan, P.; Zhang, S.J.; Jiang, X.; Wu, C.; Li, Y.; Liu, S.F.; Liu, Q.Q.; Li, J.H.; Pudasaini, B.; et al. Impact of Pituitary-Gonadal Axis Hormones on Pulmonary Arterial Hypertension in Men. Hypertension 2018, 72, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Wang, L.; Lu, W.Z.; Yuan, P.; Wu, W.H.; Zhou, Y.P.; Zhao, Q.H.; Zhang, S.J.; Li, Y.; Wu, T.; et al. Association between High FSH, Low Progesterone and Idiopathic Pulmonary Arterial Hypertension in Women of Reproductive Age. Am. J. Hypertens. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Bilan, V.; Mi, Z.; Jackson, E.K.; Schneider, F. Aromatase inhibition attenuates and ovariectomy and 4-hydroxyestradiol have mixed effects on development of angioproliferative pulmonary hypertension in female rats. Am. J. Respir. Crit. Care Med. 2013, 187, A6099. [Google Scholar]
- Mair, K.M.; Wright, A.F.; Duggan, N.; Rowlands, D.J.; Hussey, M.J.; Roberts, S.; Fullerton, J.; Nilsen, M.; Loughlin, L.; Thomas, M.; et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, A.; Nilsen, M.; Loughlin, L.; Salt, I.P.; MacLean, M.R. Metformin Reverses Development of Pulmonary Hypertension via Aromatase Inhibition. Hypertension 2016, 68, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Austin, E.D.; Talati, M.; Fesse, J.P.; Farber-Eger, E.H.; Brittain, E.L.; Hemnes, A.R.; Loyd, J.E.; West, E. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.E.; Fallon, M.B.; Krowka, M.J.; Brown, R.S.; Trotter, J.F.; Peter, I.; Tighiouart, H.; Knowles, J.A.; Rabinowitz, D.; Benza, R.L.; et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am. J. Respir. Crit. Care Med. 2009, 179, 835–842. [Google Scholar] [CrossRef]
- Kawut, S.M.; Archer-Chicko, C.L.; DeMichele, A.; Fritz, J.S.; Klinger, J.R.; Ky, B.; Palevsky, H.I.; Palmisciano, A.J.; Patel, M.; Pinder, D.; et al. Anastrozole in Pulmonary Arterial Hypertension. A Randomized, Double-Blind, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2017, 195, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Philip, J.; Vinnakota, K.C.; van den Bergh, F.; Tabima, D.M.; Hacker, T.; Beard, D.A.; Chesler, N.C. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Liu, A.; Tian, L.; Golob, M.; Eickhoff, J.C.; Boston, M.; Chesler, N.C. 17beta-Estradiol Attenuates Conduit Pulmonary Artery Mechanical Property Changes With Pulmonary Arterial Hypertension. Hypertension 2015, 66, 1082–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Schreier, D.; Tian, L.; Eickhoff, J.C.; Wang, Z.; Hacker, T.A.; Chesler, N.C. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H273–H283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umar, S.; Partow-Navid, R.; Ruffenach, G.; Iorga, A.; Moazeni, S.; Eghbali, M. Severe pulmonary hypertension in aging female apolipoprotein E-deficient mice is rescued by estrogen replacement therapy. Biol. Sex Differ. 2017, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.; Dempsie, Y.; Nilsen, M.; Wright, A.F.; Loughlin, L.; MacLean, M.R. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc. Res. 2011, 90, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, P.S.; Zhang, X.; Petrusevska, G. Progesterone inhibits vascular remodeling and attenuates monocrotaline-induced pulmonary hypertension in estrogen-deficient rats. Prilozi 2009, 30, 25–44. [Google Scholar] [PubMed]
- Blakemore, J.; Naftolin, F. Aromatase: Contributions to Physiology and Disease in Women and Men. Physiology 2016, 31, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, E.R.; Clyne, C.; Rubin, G.; Boon, W.C.; Robertson, K.; Britt, K.; Speed, C.; Jones, M. Aromatase—A brief overview. Annu. Rev. Physiol. 2002, 64, 93–127. [Google Scholar] [CrossRef]
- Simpson, E.R.; Zhao, Y.; Agarwal, V.R.; Michael, M.D.; Bulun, S.E.; Hinshelwood, M.M.; Graham-Lorence, S.; Sun, T.; Fisher, C.R.; Qin, K.; et al. Aromatase expression in health and disease. Recent Prog. Horm. Res. 1997, 52, 185–213. [Google Scholar]
- Naftolin, F.; Ryan, K.J.; Davies, I.J.; Reddy, V.V.; Flores, F.; Petro, Z.; Kuhn, M.; White, R.J.; Takaoka, Y.; Wolin, L. The formation of estrogens by central neuroendocrine tissues. Recent Prog. Horm. Res. 1975, 31, 295–319. [Google Scholar]
- Adashi, E.Y.; Hsueh, A.J. Estrogens augment the stimulation of ovarian aromatase activity by follicle-stimulating hormone in cultured rat granulosa cells. J. Biol. Chem. 1982, 257, 6077–6083. [Google Scholar]
- Purohit, A.; Singh, A.; Ghilchik, M.W.; Reed, M.J. Inhibition of tumor necrosis factor alpha-stimulated aromatase activity by microtubule-stabilizing agents, paclitaxel and 2-methoxyestradiol. Biochem. Biophys. Res. Commun. 1999, 261, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Hunger, N.I.; Docanto, M.; Simpson, E.R. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res. Treat. 2010, 123, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Bilan, V.; Jackson, E.K.; Schneider, F. Sugen 5416 Dose-Hypoxia-Normoxia-Gender Interaction in Angioproliferative Pulmonary Hypertension in Rats. Am. J. Respir. Crit. Care Med. 2014, 189, A5566. [Google Scholar]
- Dubey, R.K.; Tofovic, S.P.; Jackson, E.K. Cardiovascular pharmacology of estradiol metabolites. J. Pharmacol. Exp. Ther. 2004, 308, 403–409. [Google Scholar] [CrossRef]
- Barchiesi, F.; Jackson, E.K.; Fingerle, J.; Gillespie, D.G.; Odermatt, B.; Dubey, R.K. 2-Methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circ. Res. 2006, 99, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Docherty, C.K.; Nilsen, M.; MacLean, M.R. Influence of 2-Methoxyestradiol and Sex on Hypoxia-Induced Pulmonary Hypertension and Hypoxia-Inducible Factor-1-alpha. J. Am. Heart Assoc. 2019, 8, e011628. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Jiang, L.; Fu, C.; Wu, X.; Liu, Z.; Song, J.; Lu, H.; Wu, X.; Li, S. 2-Methoxyestradiol attenuates chronic-intermittent hypoxia-induced pulmonary hypertension through regulating microRNA-223. J. Cell Physiol. 2019, 234, 6324–6335. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, Q.; Yuan, Y.; Li, Y.; Gong, X. Effects of 17beta-estradiol and 2-methoxyestradiol on the oxidative stress-hypoxia inducible factor-1 pathway in hypoxic pulmonary hypertensive rats. Exp. Ther. Med. 2017, 13, 2537–2543. [Google Scholar] [CrossRef] [Green Version]
- Tofovic, S.P.; Jones, T.J.; Bilan, V.P.; Jackson, E.K.; Petrusevska, G. Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J. Cardiovasc. Pharmacol. 2010, 56, 475–483. [Google Scholar] [CrossRef]
- Fenoy, F.J.; Hernandez, M.E.; Hernandez, M.; Quesada, T.; Salom, M.G.; Hernandez, I. Acute effects of 2-methoxyestradiol on endothelial aortic No release in male and ovariectomized female rats. Nitric Oxide 2010, 23, 12–19. [Google Scholar] [CrossRef]
- Chen, W.; Cui, Y.; Zheng, S.; Huang, J.; Li, P.; Simoncini, T.; Zhang, Y.; Fu, X. 2-methoxyestradiol induces vasodilation by stimulating NO release via PPARgamma/PI3K/Akt pathway. PLoS ONE 2015, 10, e0118902. [Google Scholar] [CrossRef]
- Manavathi, B.; Acconcia, F.; Rayala, S.K.; Kumar, R. An inherent role of microtubule network in the action of nuclear receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 15981–15986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, S.A.; Kamel, M.W.; Botting, S.; Salih, S.M.; Borahay, M.A.; Hamed, A.A.; Kilic, G.S.; Saeed, M.; Williams, M.Y.; Diaz-Arrastia, C.R. Catechol-o-methyltransferase expression and 2-methoxyestradiol affect microtubule dynamics and modify steroid receptor signaling in leiomyoma cells. PLoS ONE 2009, 4, e7356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, S.A.; Nasr, A.B.; Dubey, R.K.; Al-Hendy, A. Estrogen metabolite 2-methoxyestradiol induces apoptosis and inhibits cell proliferation and collagen production in rat and human leiomyoma cells: A potential medicinal treatment for uterine fibroids. J. Soc. Gynecol. Investig. 2006, 13, 542–550. [Google Scholar] [CrossRef]
- Morrell, N.W.; Stenmark, K.R. The renin-angiotensin system in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 1138–1139. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.A.; Leopold, J.A. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm. Circ. 2014, 4, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.M.; Luo, L.; Guo, Z.; Yang, M.; Ye, R.S.; Luo, C. Activation of renin-angiotensin-aldosterone system (RAAS) in the lung of smoking-induced pulmonary arterial hypertension (PAH) rats. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 249–253. [Google Scholar] [CrossRef]
- Pingili, A.K.; Davidge, K.N.; Thirunavukkarasu, S.; Khan, N.S.; Katsurada, A.; Majid, D.S.A.; Gonzalez, F.J.; Navar, L.G.; Malik, K.U. 2-Methoxyestradiol Reduces Angiotensin II-Induced Hypertension and Renal Dysfunction in Ovariectomized Female and Intact Male Mice. Hypertension 2017, 69, 1104–1112. [Google Scholar] [CrossRef]
- Ueki, N.; Kanasaki, K.; Kanasaki, M.; Takeda, S.; Koya, D. Catechol-O-Methyltransferase Deficiency Leads to Hypersensitivity of the Pressor Response against Angiotensin II. Hypertension 2017, 69, 1156–1164. [Google Scholar] [CrossRef]
- Koganti, S.; Snyder, R.; Gumaste, U.; Karamyan, V.T.; Thekkumkara, T. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur. J. Pharmacol. 2014, 723, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Ogola, B.; Zhang, Y.; Iyer, L.; Thekkumkara, T. 2-Methoxyestradiol causes matrix metalloproteinase 9-mediated transactivation of epidermal growth factor receptor and angiotensin type 1 receptor downregulation in rat aortic smooth muscle cells. Am. J. Physiol. Cell Physiol. 2018, 314, C554–C568. [Google Scholar] [CrossRef] [PubMed]
- Salah, E.; Bastacky, S.I.; Jackson, E.K.; Tofovic, S.P. 2-Methoxyestradiol Attenuates Angiotensin II-Induced Hypertension, Cardiovascular Remodeling, and Renal Injury. J. Cardiovasc. Pharmacol. 2019, 73, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, G.; Wagner, R.A.; Schellong, S.; Perez, V.A.; Urashima, T.; Wang, L.; Sheikh, A.Y.; Suen, R.S.; Stewart, D.J.; Rabinovitch, M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 2007, 115, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Assad, T.R.; Hemnes, A.R. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr. Hypertens. Rep. 2015, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summer, R.; Walsh, K.; Medoff, B.D. Obesity and pulmonary arterial hypertension: Is adiponectin the molecular link between these conditions? Pulm. Circ. 2011, 1, 440–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Luther, J.M.; Rhodes, C.J.; Burgess, J.P.; Carlson, J.; Fan, R.; Fessel, J.P.; Fortune, N.; Gerszten, R.E.; Halliday, S.J.; et al. Human PAH is characterized by a pattern of lipid-related insulin resistance. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Niswender, K.D.; Johnson, J.A.; Pugh, M.E.; Gleaves, L.; Fessel, J.P.; Hemnes, A.R. A potential role for insulin resistance in experimental pulmonary hypertension. Eur. Respir. J. 2013, 41, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Annerbrink, K.; Westberg, L.; Nilsson, S.; Rosmond, R.; Holm, G.; Eriksson, E. Catechol O-methyltransferase val158-met polymorphism is associated with abdominal obesity and blood pressure in men. Metabolism 2008, 57, 708–711. [Google Scholar] [CrossRef]
- Kanasaki, M.; Srivastava, S.P.; Yang, F.; Xu, L.; Kudoh, S.; Kitada, M.; Ueki, N.; Kim, H.; Li, J.; Takeda, S.; et al. Deficiency in catechol-o-methyltransferase is linked to a disruption of glucose homeostasis in mice. Sci. Rep. 2017, 7, 7927. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.T.; Jablonski, K.A.; Chen, L.; Harden, M.; Tolkin, B.R.; Kaptchuk, T.J.; Bray, G.A.; Ridker, P.M.; Florez, J.C.; Mukamal, K.J.; et al. Catechol-O-methyltransferase association with hemoglobin A1c. Metabolism 2016, 65, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan-Lluka, L.J. Evidence for saturation of catechol-O-methyltransferase by low concentrations of noradrenaline in perfused lungs of rats. Naunyn Schmiedebergs Arch. Pharmacol. 1995, 351, 408–416. [Google Scholar] [CrossRef] [PubMed]
- De Santi, C.; Giulianotti, P.C.; Pietrabissa, A.; Mosca, F.; Pacifici, G.M. Catechol-O-methyltransferase: Variation in enzyme activity and inhibition by entacapone and tolcapone. Eur. J. Clin. Pharmacol. 1998, 54, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Boudikova, B.; Szumlanski, C.; Maidak, B.; Weinshilboum, R. Human liver catechol-O-methyltransferase pharmacogenetics. Clin. Pharmacol. Ther. 1990, 48, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xie, T.; Ramsden, D.B.; Ho, S.L. Human catechol-O-methyltransferase down-regulation by estradiol. Neuropharmacology 2003, 45, 1011–1018. [Google Scholar] [CrossRef]
- Xie, T.; Ho, S.L.; Ramsden, D. Characterization and implications of estrogenic down-regulation of human catechol-O-methyltransferase gene transcription. Mol. Pharmacol. 1999, 56, 31–38. [Google Scholar] [CrossRef]
- Parvez, S.; Parvez, S.H.; Youdim, M.B. Variation in activity of monoamine metabolizing enzymes in rat liver during pregnancy. Br. J. Pharmacol. 1975, 53, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Schendzielorz, N.; Rysa, A.; Reenila, I.; Raasmaja, A.; Mannisto, P.T. Complex estrogenic regulation of catechol-O-methyltransferase (COMT) in rats. J. Physiol. Pharmacol. 2011, 62, 483–490. [Google Scholar]
- Meyer, M.R.; Fredette, N.C.; Daniel, C.; Sharma, G.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. Obligatory role for GPER in cardiovascular aging and disease. Sci. Signal. 2016, 9, ra105. [Google Scholar] [CrossRef] [Green Version]
- Alencar, A.K.; Montes, G.C.; Montagnoli, T.; Silva, A.M.; Martinez, S.T.; Fraga, A.G.; Wang, H.; Groban, L.; Sudo, R.T.; Zapata-Sudo, G. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur. J. Pharm. Sci. 2017, 97, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Alencar, A.K.; Montes, G.C.; Costa, D.G.; Mendes, L.V.; Silva, A.M.; Martinez, S.T.; Trachez, M.M.; Cunha, V.D.M.; Montagnoli, T.L.; Fraga, A.G.; et al. Cardioprotection Induced by Activation of GPER in Ovariectomized Rats With Pulmonary Hypertension. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaufelberger, S.A.; Rosselli, M.; Barchiesi, F.; Gillespie, D.G.; Jackson, E.K.; Dubey, R.K. 2-Methoxyestradiol, an endogenous 17beta-estradiol metabolite, inhibits microglial proliferation and activation via an estrogen receptor-independent mechanism. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E313–E322. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Jones, T.; Petrusevska, G. Dose-dependent therapeutic effects of 2-Methoxyestradiol on Monocrotaline-Induced pulmonary hypertension and vascular remodeling. Prilozi 2010, 31, 279–295. [Google Scholar] [PubMed]
- Tofovic, S.P.; Zhang, X.C.; Jones, T.; Jackson, E.K.; Petrusevska, G. 2-Methoxyestradiol attenuates the development and retards the progression of chronic hypoxia-induced pulmonary hypertension in rats. Circulation 2005, 112, 98–99. [Google Scholar]
- Tofovic, S.P.; Jackson, E.K.; Rafikova, O. Synergistic Effects of 2-Methoxyestradiol and Retinoic Acid on Amelioration of Monocrotaline-Induced Pulmonary Hypertension. Am. J. Res. Crit. Care Med. 2008, 177, A592. [Google Scholar]
- Tofovic, S.P.; Zhu, H.; Jackson, E.K.; Rafikova, O. 2-Methoxyestrone Inhibits Vascular Remodeling and Attenuates Monocrotaline-Induced Pulmonary Hypertension. Eur. Res. J. 2008, 32 (Suppl. 52), 995. [Google Scholar]
- Hu, J.; Rafikova, O.; Schneider, F.; Jackson, E.K. Tofovic SP 2-Methoxyestrone Retards the Progression of Angioproliferative Pulmonary Hypertension in Rats—Role of 17beta-Hydroxysteroid Dehydrogenase Pathway. Circulation 2015, 132 (Suppl. 3), A18888. [Google Scholar]
- Tofovic, S.P.; Hu, J.; Jackson, E.K.; Schneider, F. 2-Hydroxyestradiol Attenuates Metabolic Syndrome-Induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2015, 191, A4096. [Google Scholar]
- White, K.; Johansen, A.K.; Nilsen, M.; Ciuclan, L.; Wallace, E.; Paton, L.; Campbell, A.; Morecroft, I.; Loughlin, L.; McClure, J.D.; et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 2012, 126, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Talati, M.; Fessel, J.P.; Hemnes, A.R.; Gladson, S.; French, J.; Shay, S.; Trammell, A.; Phillips, J.A.; Hamid, R.; et al. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation 2016, 133, 82–97. [Google Scholar] [CrossRef]
- Swaneck, G.E.; Fishman, J. Covalent binding of the endogenous estrogen 16 alpha-hydroxyestrone to estradiol receptor in human breast cancer cells: Characterization and intranuclear localization. Proc. Natl. Acad. Sci. USA 1988, 85, 7831–7835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Cogan, J.D.; West, J.D.; Hedges, L.K.; Hamid, R.; Dawson, E.P.; Wheeler, L.A.; Parl, F.F.; Loyd, J.E.; Phillips, J. Alterations in oestrogen metabolism: Implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur. Respir. J. 2009, 34, 1093–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyd, J.E. Pulmonary arterial hypertension: Insights from genetic studies. Proc. Am. Thorac. Soc. 2011, 8, 154–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.; Cogan, J.; Geraci, M.; Robinson, L.; Newman, J.; Phillips, J.A.; Lane, K.; Meyrick, B.; Loyd, J. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med. Genom. 2008, 1, 45. [Google Scholar] [CrossRef] [Green Version]
- Johansen, A.K.; Dean, A.; Morecroft, I.; Hood, K.; Nilsen, M.; Loughlin, L.; Anagnostopoulou, A.; Touyz, R.M.; White, K.; MacLean, M.R. The serotonin transporter promotes a pathological estrogen metabolic pathway in pulmonary hypertension via cytochrome P450 1B1. Pulm. Circ. 2016, 6, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16alpha-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef]
- Ventetuolo, C.E.; Mitra, N.; Wan, F.; Manichaikul, A.; Barr, R.G.; Johnson, C.; Bluemke, D.A.; Lima, J.A.; Tandri, H.; Ouyang, P.; et al. Oestradiol metabolism and androgen receptor genotypes are associated with right ventricular function. Eur. Respir. J. 2016, 47, 553–563. [Google Scholar] [CrossRef]
- Kerzee, J.K.; Ramos, K.S. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: Role of the Ahr bHLH/PAS transcription factor. Circ. Res. 2001, 89, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef]
- Rahman, M.; Sutter, C.H.; Emmert, G.L.; Sutter, T.R. Regioselective 2-hydroxylation of 17beta-estradiol by rat cytochrome P4501B1. Toxicol. Appl. Pharmacol. 2006, 216, 469–478. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dawling, S.; Roodi, N.; Parl, F.F. Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res. 2003, 63, 3127–3132. [Google Scholar] [PubMed]
- Maayah, Z.H.; Levasseur, J.; Piragasam, R.S.; Abdelhamid, G.; Dyck, J.R.B.; Fahlman, R.P.; Siraki, A.G.; El-Kadi, A.O.S. 2-Methoxyestradiol protects against pressure overload-induced left ventricular hypertrophy. Sci. Rep. 2018, 8, 2780. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, D.; Jansson, I.; Stoilov, I.; Sarfarazi, M.; Schenkman, J.B. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab. Dispos. 2004, 32, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Parrish, A.R.; Ramos, K.S. Constitutive and inducible expression of cytochrome P450IA1 and P450IB1 in human vascular endothelial and smooth muscle cells. In Vitro Cell. Dev. Biol. Anim. 1998, 34, 671–673. [Google Scholar] [CrossRef]
- Zhu, D.; Ran, Y. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J. Physiol. Sci. 2012, 62, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, S.; Wang, Z.; Ma, J.; Liu, S.; Li, W.; Zhu, D. The role of ERK1/2 in 15-HETE-inhibited apoptosis in pulmonary arterial smooth muscle cells. J. Recept. Signal. Transduct. Res. 2011, 31, 45–52. [Google Scholar] [CrossRef]
- Sugumaran, P.K.; Wang, S.; Song, S.; Nie, X.; Zhang, L.; Feng, Y.; Ma, W.; Zhu, D. 15-oxo-Eicosatetraenoic acid prevents serum deprivation-induced apoptosis of pulmonary arterial smooth muscle cells by activating pro-survival pathway. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 89–98. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Althurwi, H.N.; Abdelhamid, G.; Lesyk, G.; Jurasz, P.; El-Kadi, A.O. CYP1B1 inhibition attenuates doxorubicin-induced cardiotoxicity through a mid-chain HETEs-dependent mechanism. Pharmacol. Res. 2016, 105, 28–43. [Google Scholar] [CrossRef]
- Ma, C.; Li, Y.; Ma, J.; Liu, Y.; Li, Q.; Niu, S.; Shen, Z.; Zhang, L.; Pan, Z.; Zhu, D. Key role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension 2011, 58, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Al-Naamani, N.; Sagliani, K.D.; Dolnikowski, G.G.; Warburton, R.R.; Toksoz, D.; Kayyali, U.; Hill, N.S.; Fanburg, B.L.; Roberts, K.E.; Preston, I.R. Plasma 12- and 15-hydroxyeicosanoids are predictors of survival in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Sun, D. Sexually Dimorphic Regulation of EET Synthesis and Metabolism: Roles of Estrogen. Front. Pharmacol. 2018, 9, 1222. [Google Scholar] [CrossRef] [PubMed]
- Somjen, D.; Kohen, F.; Limor, R.; Sharon, O.; Knoll, E.; Many, A.; Stern, N. Estradiol-17beta increases 12- and 15-lipoxygenase (type2) expression and activity and reactive oxygen species in human umbilical vascular smooth muscle cells. J. Steroid Biochem. Mol. Biol. 2016, 163, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandhi, S.; Qin, J.; Froogh, G.; Jiang, H.; Luo, M.; Wolin, M.S.; Huang, A.; Sun, D. EET-dependent potentiation of pulmonary arterial pressure: Sex-different regulation of soluble epoxide hydrolase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1478–L1486. [Google Scholar] [CrossRef] [Green Version]
- Kandhi, S.; Zhang, B.; Froogh, G.; Qin, J.; Alruwaili, N.; Le, Y.; Yang, Y.M.; Hwang, S.H.; Hammock, B.D.; Wolin, M.S.; et al. EETs promote hypoxic pulmonary vasoconstriction via constrictor prostanoids. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L350–L359. [Google Scholar] [CrossRef]
- Keseru, B.; Barbosa-Sicard, E.; Schermuly, R.T.; Tanaka, H.; Hammock, B.D.; Weissmann, N.; Fisslthaler, B.; Fleming, I. Hypoxia-induced pulmonary hypertension: Comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc. Res. 2010, 85, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, C.L.; Vesell, E.S. Hormonal regulation of the developmental pattern of epoxide hydrolases. Studies in rat liver. Biochem. Pharmacol. 1989, 38, 603–610. [Google Scholar] [CrossRef]
- Pinot, F.; Grant, D.F.; Spearow, J.L.; Parker, A.G.; Hammock, B.D. Differential regulation of soluble epoxide hydrolase by clofibrate and sexual hormones in the liver and kidneys of mice. Biochem. Pharmacol. 1995, 50, 501–508. [Google Scholar] [CrossRef]
- Bayard, F.; Clamens, S.; Meggetto, F.; Blaes, N.; Delsol, G.; Faye, J.C. Estrogen synthesis, estrogen metabolism, and functional estrogen receptors in rat arterial smooth muscle cells in culture. Endocrinology 1995, 136, 1523–1529. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Chen, J.; Yin, D.C.; Lin, S.X. The contribution of 17beta-hydroxysteroid dehydrogenase type 1 to the estradiol-estrone ratio in estrogen-sensitive breast cancer cells. PLoS ONE 2012, 7, e29835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubhye, J.; Alard, I.C.; van Antwerpen, P.; Dufrasne, F. Type 2 17-beta hydroxysteroid dehydrogenase as a novel target for the treatment of osteoporosis. Future Med. Chem. 2015, 7, 1431–1456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Lee, W.J.; Zhu, B.T. Selective insensitivity of ZR-75-1 human breast cancer cells to 2-methoxyestradiol: Evidence for type II 17beta-hydroxysteroid dehydrogenase as the underlying cause. Cancer Res. 2005, 65, 5802–5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, K.; Okouneva, T.; Larson, G.; Panda, D.; Wilson, L.; Jordan, M.A. 2-Methoxyestradiol suppresses microtubule dynamics and arrests mitosis without depolymerizing microtubules. Mol. Cancer Ther. 2006, 5, 2225–2233. [Google Scholar] [CrossRef] [Green Version]
- James, J.; Murry, D.J.; Treston, A.M.; Storniolo, A.M.; Sledge, G.W.; Sidor, C.; Miller, K.D. Phase I safety, pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol alone or in combination with docetaxel in patients with locally recurrent or metastatic breast cancer. Investig. New Drugs 2007, 25, 41–48. [Google Scholar] [CrossRef]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef]
- Sweeney, C.; Liu, G.; Yiannoutsos, C.; Kolesar, J.; Horvath, D.; Staab, M.J.; Fife, K.; Armstrong, V.; Treston, A.; Sidor, C.; et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin. Cancer Res. 2005, 11, 6625–6633. [Google Scholar] [CrossRef] [Green Version]
- Cool, C.D.; Stewart, J.S.; Werahera, P.; Miller, G.J.; Williams, R.L.; Voelkel, N.F.; Tuder, R.M. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers: Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am. J. Pathol. 1999, 155, 411–419. [Google Scholar] [CrossRef]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.L.; Law, T.C. Chronic hypoxia- and monocrotaline-induced elevation of hypoxia-inducible factor-1 alpha levels and pulmonary hypertension. J. Biomed. Sci. 2004, 11, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Lu, L.; Wang, L.; Chen, M.; Hu, T. Expression and Correlation of Hypoxia-Inducible Factor-1alpha (HIF-1alpha) with Pulmonary Artery Remodeling and Right Ventricular Hypertrophy in Experimental Pulmonary Embolism. Med. Sci. Monit. 2017, 23, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Concina, P.; Sordello, S.; Barbacanne, M.A.; Elhage, R.; Pieraggi, M.T.; Fournial, G.; Plouet, J.; Bayard, F.; Arnal, J.F. The mitogenic effect of 17beta-estradiol on in vitro endothelial cell proliferation and on in vivo reendothelialization are both dependent on vascular endothelial growth factor. J. Vasc. Res. 2000, 37, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.E.; McGowan, K.A.; Grant, D.S.; Maheshwari, S.; Bhartiya, D.; Cid, M.C.; Kleinman, H.K.; Schnaper, H.W. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 1995, 91, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofovic, S.P.; Jackson, E.K. 2-Methoxyestradiol in Pulmonary Arterial Hypertension: A New Disease Modifier. In Interventional Pulmonology and Pulmonary Hypertension; Intec Open: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Escuin, D.; Kline, E.R.; Giannakakou, P. Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1alpha accumulation and activity by disrupting microtubule function. Cancer Res. 2005, 65, 9021–9028. [Google Scholar] [CrossRef] [Green Version]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 2010, 176, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Basini, G.; Grasselli, F.; Bussolati, S.; Baioni, L.; Bianchi, F.; Musci, M.; Careri, M.; Mangia, A. Hypoxia stimulates the production of the angiogenesis inhibitor 2-methoxyestradiol by swine granulosa cells. Steroids 2011, 76, 1433–1436. [Google Scholar] [CrossRef]
- Plum, S.M.; Park, E.J.; Strawn, S.J.; Moore, E.G.; Sidor, C.F.; Fogler, W.E. Disease modifying and antiangiogenic activity of 2-methoxyestradiol in a murine model of rheumatoid arthritis. BMC Musculoskelet. Disord. 2009, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Philip, J.L.; Tabima, D.M.; Wolf, G.D.; Frump, A.L.; Cheng, T.C.; Schreier, D.A.; Hacker, T.A.; Lahm, T.; Chesler, N.C. Exogenous Estrogen Preserves Distal Pulmonary Arterial Mechanics and Prevents Pulmonary Hypertension in Rats. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuan, T.; Zhang, H.; Yan, Y.; Wang, D.; Fang, L.; Lu, Y.; Du, G. Activation of Nrf2 Attenuates Pulmonary Vascular Remodeling via Inhibiting Endothelial-to-Mesenchymal Transition: An Insight from a Plant Polyphenol. Int. J. Biol. Sci. 2017, 13, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [PubMed] [Green Version]
- Hopper, R.K.; Moonen, J.R.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Frid, M.; Perros, F. Endothelial-to-Mesenchymal Transition: An Evolving Paradigm and a Promising Therapeutic Target in PAH. Circulation 2016, 133, 1734–1737. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, X.; Lu, J.; Zhu, L.; Li, M. Autophagy mediates 2-methoxyestradiol-inhibited scleroderma collagen synthesis and endothelial-to-mesenchymal transition induced by hypoxia. Rheumatology 2019, 58, 1966–1975. [Google Scholar] [CrossRef]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A Hypoxia-Induced Vascular Endothelial-to-Mesenchymal Transition in Development of Radiation-Induced Pulmonary Fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.; Soro-Arnaiz, I.; de Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yan, J.; Shen, Y.; Liu, Y.; Ma, Z. Dichloroacetate prevents but not reverses the formation of neointimal lesions in a rat model of severe pulmonary arterial hypertension. Mol. Med. Rep. 2014, 10, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, S.A.; Mohammad, M.A.; Diaz-Arrastia, C.R.; Kamel, M.W.; Kilic, G.S.; Ndofor, B.T.; Abdel-Baki, M.S.; Theiler, S.K. Estradiol-17beta upregulates pyruvate kinase M2 expression to coactivate estrogen receptor-alpha and to integrate metabolic reprogramming with the mitogenic response in endometrial cells. J. Clin. Endocrinol. Metab. 2014, 99, 3790–3799. [Google Scholar] [CrossRef] [Green Version]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Ferri, N.; Cignarella, A.; Trevisi, L.; Bolego, C. The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1. J. Pharmacol. Exp. Ther. 2017, 361, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Jiang, H.; Li, Z.; Zhuang, Y.; Liu, Y.; Zhou, S.; Xiao, Y.; Xie, C.; Zhou, F.; Zhou, Y. 2-Methoxyestradiol enhances radiosensitivity in radioresistant melanoma MDA-MB-435R cells by regulating glycolysis via HIF-1alpha/PDK1 axis. Int. J. Oncol. 2017, 50, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Kuebler, W.M.; Bonnet, S.; Tabuchi, A. Inflammation and autoimmunity in pulmonary hypertension: Is there a role for endothelial adhesion molecules? (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893218757596. [Google Scholar] [CrossRef] [Green Version]
- Nicolls, M.R.; Voelkel, N.F. The Roles of Immunity in the Prevention and Evolution of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cracowski, J.L.; Chabot, F.; Labarere, J.; Faure, P.; Degano, B.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; Cracowski, C.; Sitbon, O.; et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 915–917. [Google Scholar] [CrossRef] [Green Version]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdiarmid, F.; Wang, D.; Duncan, L.J.; Purohit, A.; Ghilchick, M.W.; Reed, M.J. Stimulation of aromatase activity in breast fibroblasts by tumor necrosis factor alpha. Mol. Cell. Endocrinol. 1994, 106, 17–21. [Google Scholar] [CrossRef]
- Seriolo, B.; Accardo, S.; Garnero, A.; Fasciolo, D.; Cutolo, M. Association between anticardiolipin antibody positivity and increased 17-beta-estradiol levels in premenopausal women with rheumatoid arthritis. Ann. N. Y. Acad. Sci. 1999, 876, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, S.H.; Ahmari, S.E.; Bryan-Poole, N.; Evans, G.F.; Short, L.; Glasebrook, A.L. Estriol: A potent regulator of TNF and IL-6 expression in a murine model of endotoxemia. Inflammation 1996, 20, 581–597. [Google Scholar] [CrossRef]
- Foo, Y.Z.; Nakagawa, S.; Rhodes, G.; Simmons, L.W. The effects of sex hormones on immune function: A meta-analysis. Biol. Rev. 2017, 92, 551–571. [Google Scholar] [CrossRef] [Green Version]
- Roved, J.; Westerdahl, H.; Hasselquist, D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm. Behav. 2017, 88, 95–105. [Google Scholar] [CrossRef]
- Osman, M.S.; Michelakis, E.D. Immunity Comes to Play in the “Sex Paradox” of Pulmonary Arterial Hypertension. Circ. Res. 2018, 122, 1635–1637. [Google Scholar] [CrossRef]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Vaananen, K.; Watford, W.T.; Chen, Z. Estrogen receptor alpha contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folomeev, M.; Dougados, M.; Beaune, J.; Kouyoumdjian, J.C.; Nahoul, K.; Amor, B.; Alekberova, Z. Plasma sex hormones and aromatase activity in tissues of patients with systemic lupus erythematosus. Lupus 1992, 1, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tengstrand, B.; Carlstrom, K.; Fellander-Tsai, L.; Hafstrom, I. Abnormal levels of serum dehydroepiandrosterone, estrone, and estradiol in men with rheumatoid arthritis: High correlation between serum estradiol and current degree of inflammation. J. Rheumatol. 2003, 30, 2338–2343. [Google Scholar] [PubMed]
- Capellino, S.; Straub, R.H.; Cutolo, M. Aromatase and regulation of the estrogen-to-androgen ratio in synovial tissue inflammation: Common pathway in both sexes. Ann. N. Y. Acad. Sci. 2014, 1317, 24–31. [Google Scholar] [CrossRef]
- Shand, F.H.; Langenbach, S.Y.; Keenan, C.R.; Ma, S.P.; Wheaton, B.J.; Schuliga, M.J.; Ziogas, J.; Stewart, A.G. In vitro and in vivo evidence for anti-inflammatory properties of 2-methoxyestradiol. J. Pharmacol. Exp. Ther. 2011, 336, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, T.E.; Anderson, R.L.; Hughes, R.A.; Altmann, E.; Schuliga, M.; Ziogas, J.; Stewart, A.G. 2-Methoxyestradiol—A unique blend of activities generating a new class of anti-tumour/anti-inflammatory agents. Drug Discov. Today 2007, 12, 577–584. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Dubey, R.K.; Jackson, E.K. 2-Hydroxyestradiol attenuates the development of obesity, the metabolic syndrome, and vascular and renal dysfunction in obese ZSF1 rats. J. Pharmacol. Exp. Ther. 2001, 299, 973–977. [Google Scholar]
- Tofovic, S.P.; Salah, E.M.; Dubey, R.K.; Melhem, M.F.; Jackson, E.K. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic nitric oxide synthase inhibition. J. Cardiovasc. Pharmacol. 2005, 46, 25–35. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, Y.; Jackson, E.K.; Tofovic, S.P. 2-Methoxyestradiol and 2-ethoxyestradiol retard the progression of renal disease in aged, obese, diabetic ZSF1 rats. J. Cardiovasc. Pharmacol. 2007, 49, 56–63. [Google Scholar] [CrossRef]
- Duncan, G.S.; Brenner, D.; Tusche, M.W.; Brustle, A.; Knobbe, C.B.; Elia, A.J.; Mock, T.; Bray, M.R.; Krammer, P.H.; Mak, T.W. 2-Methoxyestradiol inhibits experimental autoimmune encephalomyelitis through suppression of immune cell activation. Proc. Natl. Acad. Sci. USA 2012, 109, 21034–21039. [Google Scholar] [CrossRef] [Green Version]
- Luc, J.G.; Paulin, R.; Zhao, J.Y.; Freed, D.H.; Michelakis, E.D.; Nagendran, J. 2-Methoxyestradiol: A Hormonal Metabolite Modulates Stimulated T-Cells Function and proliferation. Transplant. Proc. 2015, 47, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, T.; Su, S.; Wang, F. 2-Methoxyestradiol Alleviates Experimental Autoimmune Uveitis by Inhibiting Lymphocytes Proliferation and T cell Differentiation. BioMed Res. Int. 2016, 2016, 7948345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, B.D.; Shtraichman, O.; Langleben, D.; Shimony, A.; Kramer, M.R. Combination Therapy for Pulmonary Arterial Hypertension: A Systematic Review and Meta-analysis. Can. J. Cardiol. 2016, 32, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, A.C.; Bonnet, S.; Provencher, S. Combination therapy in pulmonary arterial hypertension: Recent accomplishments and future challenges. Pulm. Circ. 2017, 7, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
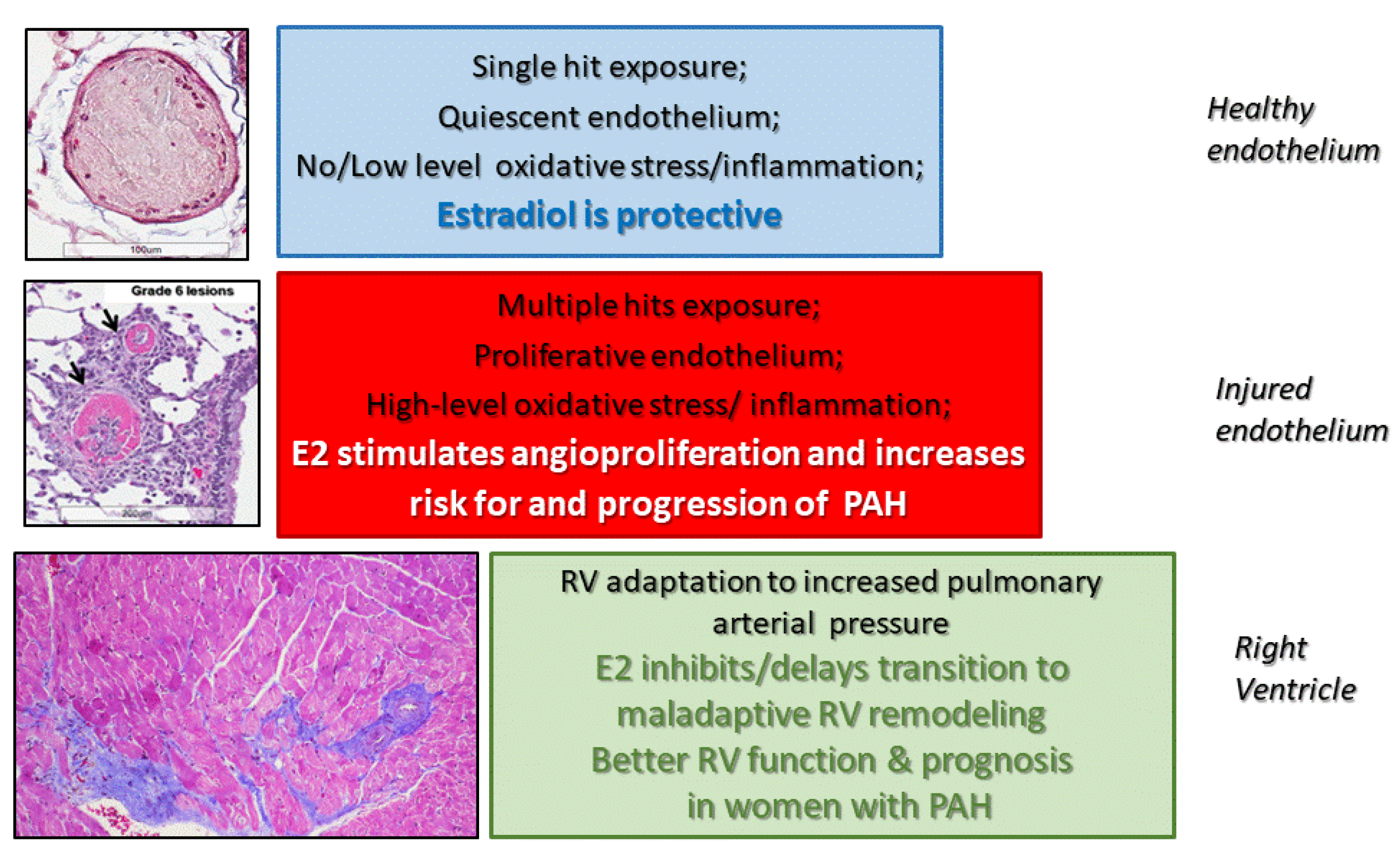{kind=link}
| Animal Model PAH Patients | Hormone, Sex, Enzyme, Treatment | Effects/Comments | References |
|---|---|---|---|
| CH- and MCT-PH in rats | Estradiol (E2) | Female sex protective; E2 attenuate PH and RV and vascular remodeling; OVX exacerbates PH | Reviewed previously in Tofovic SP, 2010 [8] |
| MCT female and OVX rats | Estradiol | E2 attenuates whereas OVX exacerbates PH; ↓ E2 levels and aromatase, ↑ Cyp 1A1 and Cyp1B1 | Yuan P. et al., 2013 [48] |
| Su + Hx male and female rats | Female versus Male | Female: greater pulmonary vascular remodeling; Male: more severe RV failure, lower survival rate | Rafikova et al., 2015 [49] |
| Chronic hypoxia (CH)- induced PH in rats | DHEA | Prevents or reverses PH; Inhibits PH and augments vasodilator responsiveness to NO | Bonnet S. et al., 2003; Oka M. et al., 2007 [37,39] |
| MCT + pneumonectomy rat | DHEA | Rescue treatment attenuates PH and vascular remodeling and eliminates late mortality | Homma N. et al., 2008 [40] |
| MCT rat | DHEA | Reduces PH and RV hypertrophy | Paulin R. et al., 2011 [41] |
| Su + Hx rat | DHEA | Attenuates PH; restores RV structure/function | Alzoubi A. et al., 2012 [44] |
| CH-PH rats Rat-pup model of CH-PH | DHEA | Reverses CH/reoxygenation-induced RV dysfunction; Attenuates PH, RV, and vascular remodeling in infant rats | Dumas de La Roque E. et al., 2012; 2013 [43,50] |
| Pregnant Sheep | DHEA | Vasodilator effects on fetal pulmonary circulation | Sharma D. et al., 2018 [42] |
| Rat model of left heart failure (LHF)-induced PH | DHEA | Attenuates LHF-induced PH and RV and vascular remodeling | Zhang YT. et al., 2019 [51] |
| PM women, idiopathic, CTD- or CHD-PAH | Low DHEA-S High E2 | Increased risk and severity of PAH in postmenopausal women | Baird GL et al., 2018 [47] |
| Men with idiopathic, heritable, or CTD-PAH | Low DHEA-S High E2 | Associated with risk of PAH; ↑ E2, shorter 6MWD; ↑ DHEA-S, lower right atrial pressure | Ventetuolo CE et al., 2016 [46] |
| Patients with COPD | DHEA | Improves PH in COPD patients | D. La Roque E. et al., 2012 [45] |
| Men with idiopathic PAH | Estradiol Testosterone (T) Progesterone (P) | ↑ E2 and E2/T ratio and ↓ T and P associated with ↑ risk of PAH; high E2 independently associated with higher mortality | Wu W-H. et al., 2018 [52] |
| Premenopausal women with idiopathic PAH | FSH Progesterone | ↑ FSH and ↓ P tended to be associated with high risk of IPAH and mortality among patients | Zhang Y-X et al., 2019 [53] |
| Su+Hx female rats | Anastrozole | Reduces PH and number of occlusive and PLX lesions, but has no effect on RV remodeling. | Tofovic SP. et al., 2013 [54] * |
| CH mice and Su+Hx rats | Anastrozole | Attenuates PH only in female animals | Mair KM. et al., 2014 [55] |
| Su+Hx rats | Metformin | ↓ circulating estrogens and aromatase levels/activity, and attenuates PH and vascular and RV remodeling | Dean A. et al., 2016 [56] |
| BMPR2 mutant mice | Anastrozole + Fulvestrant | Prevents and Reverses PH and reduces BMPR2 mutation associated with metabolic defects | Chen X. et al., 2017 [57] |
| Portopulmonary hypertension | Estradiol Aromatase | Irrespective of gender, in patients with liver disease, ↑ aromatase and ↑ E2 levels increase risk of portopulmonary PH | Roberts KE. et al., 2009 [58] |
| PAH patients | Anastrozole | Reduces serum E2 and E1 levels, significantly increases 6MWD, but has no effect on RV function | Kawut SM et al., 2017 [59] |
| Su + Hx OVX female rats | Estradiol | Protects RV function and restores RV ventricular–vascular coupling efficiency | Liu A. et al., 2017 [60] |
| Su + Hx male, female and OVX rats | Estradiol | Endogenous and exogenous E2 exerts protective effects on baseline RV function and after an acute exercise challenge | Frump, AL. et al., 2015 [15] Lahm T. et al., 2016 [13] |
| Su + Hx female and OVX mice | Estradiol | E2 has no effect on PH, yet reduces proximal conduit arteries stiffness and improves ventricular–vascular coupling; no data on RV remodeling available | Liu A. et al., 2015 [61] |
| Su + Hx female and OVX mice | Estradiol | In absence of RV remodeling in diseased animals, improves RV function (enhances RV contractility in response to PH and preserves cardiac reserve) | Liu A. et al., 2014 [62] |
| MCT in Apo E deficient female mice | Estradiol | Rescue treatment reduces PH and RV hypertrophy | Umar S. et al., 2017 [63] |
| Female and OVX mice overexpressing SERT | Estradiol | Only female, SERT+ mice develop PAH, OVX abolishes and E2 treatment of OVX + SERT mice reestablishes PH. | White K. et al., 2011 [64] |
| MCT OVX rats | Progesterone | Attenuates PH, and RV and vascular remodeling | Tofovic SP. et al., 2009 [65] |
| Animal Model | Estradiol Metabolite | Effects Comments | Reference |
|---|---|---|---|
| MCT male rats | 2-methoxyestradiol 2-hydroxyestardiol | Attenuate development or progression of PH | Tofovic SP et al., 2005 [12] |
| MCT OVX and female rats | 2-methoxyestradiol | Ovariectomy exacerbates PH, whereas 2ME attenuates PH in OVX rats; no estrogenic effects | Tofovic SP et al., 2006 [26] |
| MCT male rats | 2-ethoxyestradiol | Synthetic metabolite; attenuates PH, lung inflammation, and RV and vascular modeling, and eliminates late mortality | Tofovic SP et al., 2008 [22] |
| Bleomycin-induced PH and lung fibrosis, OVX female rats | 2-methoxyestradiol | Ovariectomy exacerbates whereas 2ME attenuates PH, inflammation, fibrosis and vascular remodeling | Tofovic SP et al., 2009 [21] |
| MCT male rats | 2-methoxyestradiol | Dose-dependent therapeutic effects on PH, lung inflammation and RV and vascular remodeling; no estrogenic effects | Tofovic SP et al., 2010 [113] |
| MCT male rats | 2-methoxyestradiol | Synergistic effects with bosentan or sildenafil on amelioration of PH, lung inflammation, and vascular remodeling | Tofovic SP et al., 2010 [12] |
| CH male and female rats | 2-methoxyestradiol | Attenuates development and retards the progression of PH; decreases HIF-1α expression and reduces elevated hematocrit | Wang L. et al., 2017; Hao S. et al., 2019 Docherty CK. et al., 2019, Tofovic SP. et al., 2005 * [77,78,79,114] |
| MCT male rats | 2-methoxyestradiol +/− retinoic acid | Synergistic effects of 2ME with retinoic acid on amelioration of PH | Tofovic SP et al., 2008 [115] * |
| MCT OVX rats; Female and OVX Su+Hx rats | 2-methoxyestrone | Attenuates MCT-induced PH, inflammation and RV and vascular remodeling; attenuates Su+Hx-induced PH, RV dysfunction, and remodeling and number of occlusive lesions | Tofovic SP. et al., 2008, Hu J. et al., 2016 [116,117] * |
| Su+Hx OVX rats | 4-hydroxyestradiol | Has no effect on PH, but attenuates RV hypertrophy | Tofovic SP. et al., 2013 [54] * |
| Obese ZDSD rats | 2-hydroxyestradiol | Reduces HbA1c and attenuates metabolic syndrome-induced PH in male rats | Tofovic SP. et al., 2014 [118] * |
| Female mice | 16α - hydroxyestrone | Induces mild PH and RV and vascular remodeling | White K. et al., 2012 [119] |
| BMPR2 mutant mice | 16α -hydroxyestrone | Exacerbates BMPR2-associated PH | Chen X. et al., 2016 [120] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tofovic, S.P.; Jackson, E.K. Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 116. https://doi.org/10.3390/ijms21010116
Tofovic SP, Jackson EK. Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2020; 21(1):116. https://doi.org/10.3390/ijms21010116
Chicago/Turabian StyleTofovic, Stevan P., and Edwin K. Jackson. 2020. "Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 21, no. 1: 116. https://doi.org/10.3390/ijms21010116
APA StyleTofovic, S. P., & Jackson, E. K. (2020). Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 21(1), 116. https://doi.org/10.3390/ijms21010116