Direct Sarcomere Modulators Are Promising New Treatments for Cardiomyopathies


Abstract
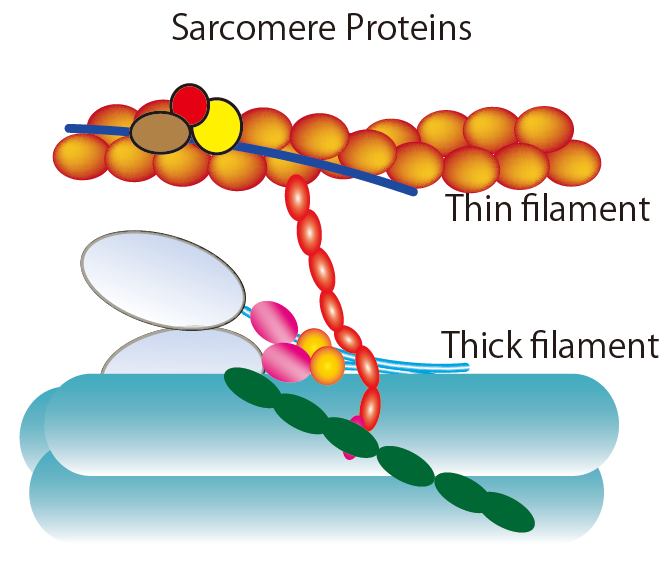
:
{kind=link}
{kind=link}
{kind=link}
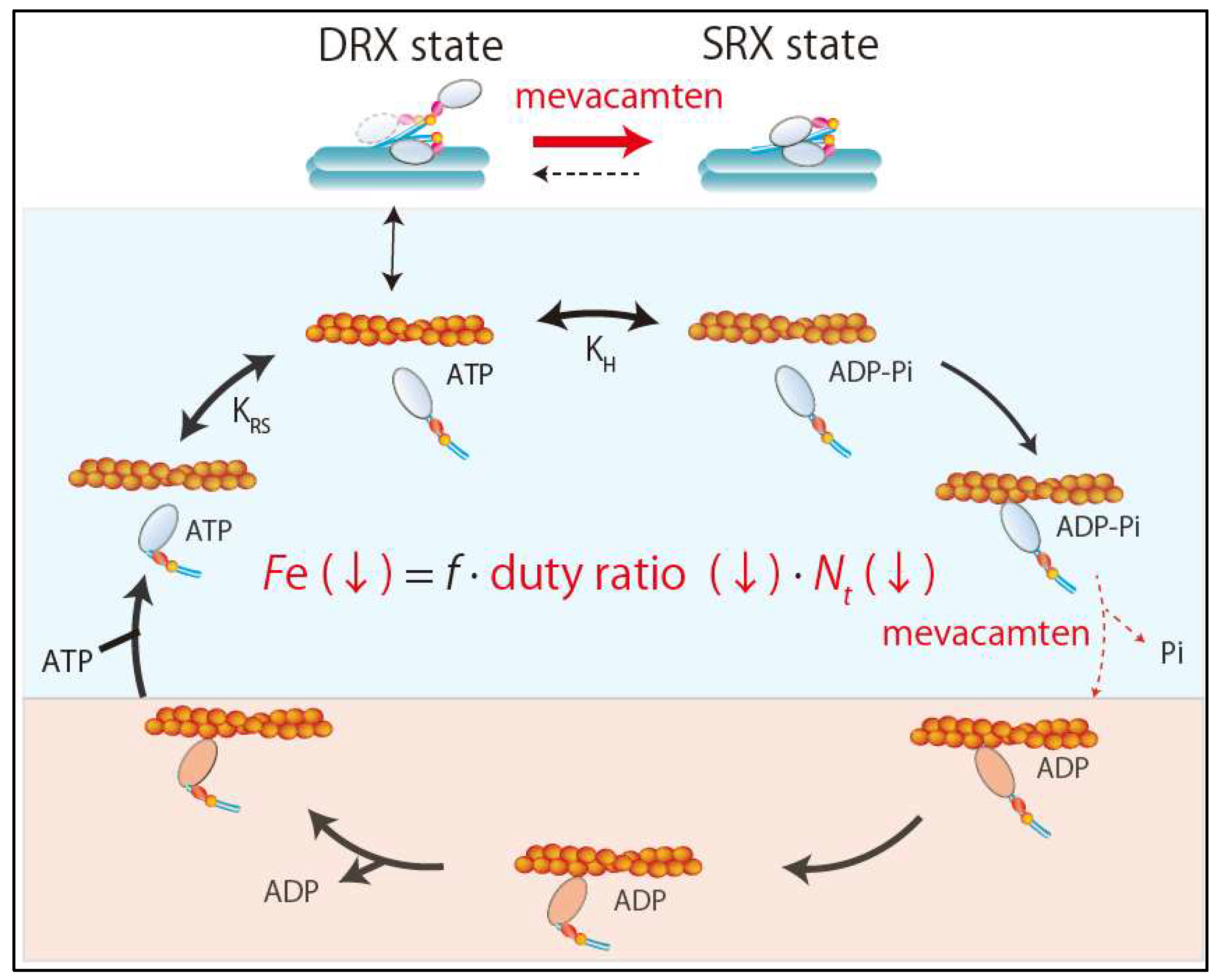{kind=link}
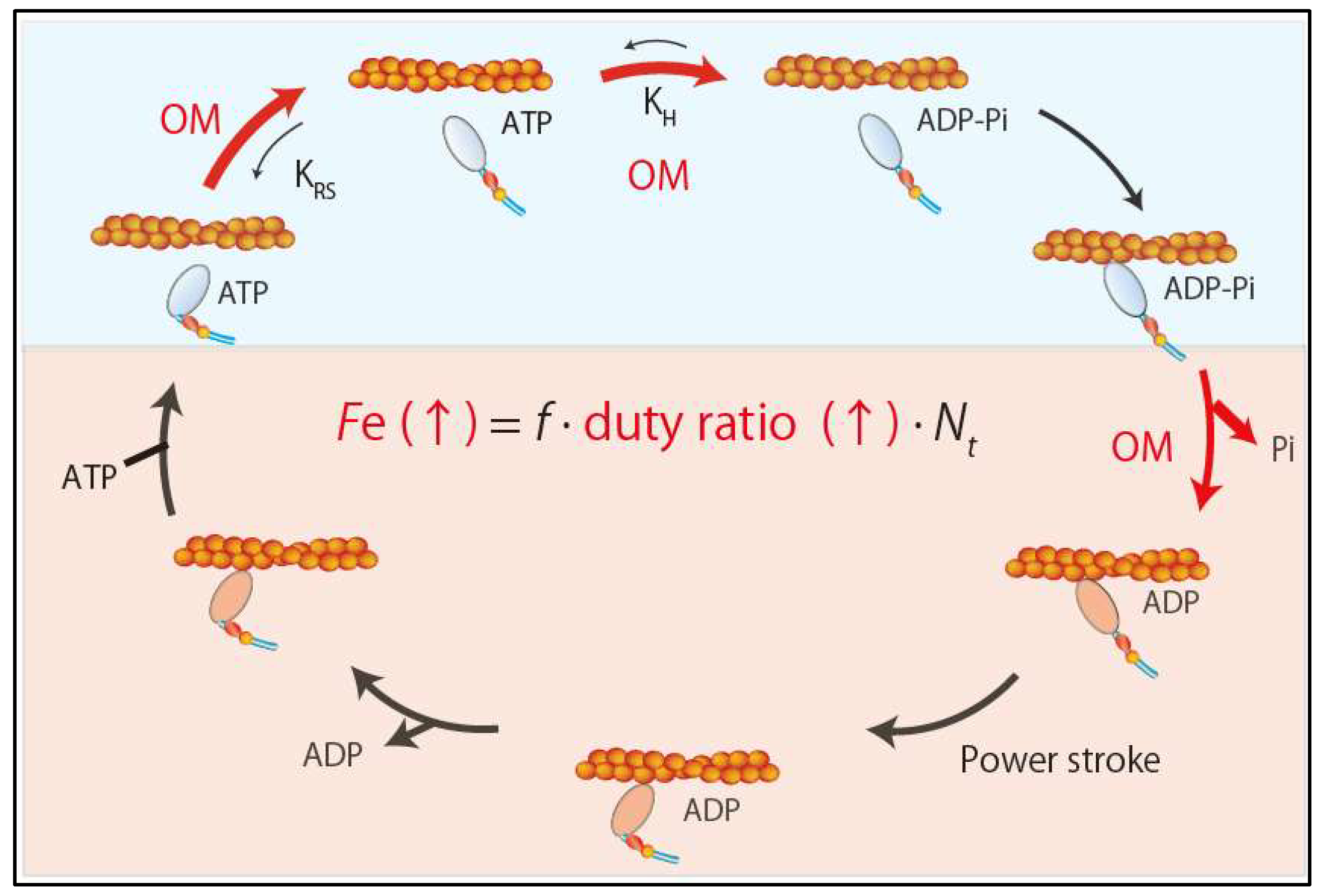{kind=link}
1. Introduction
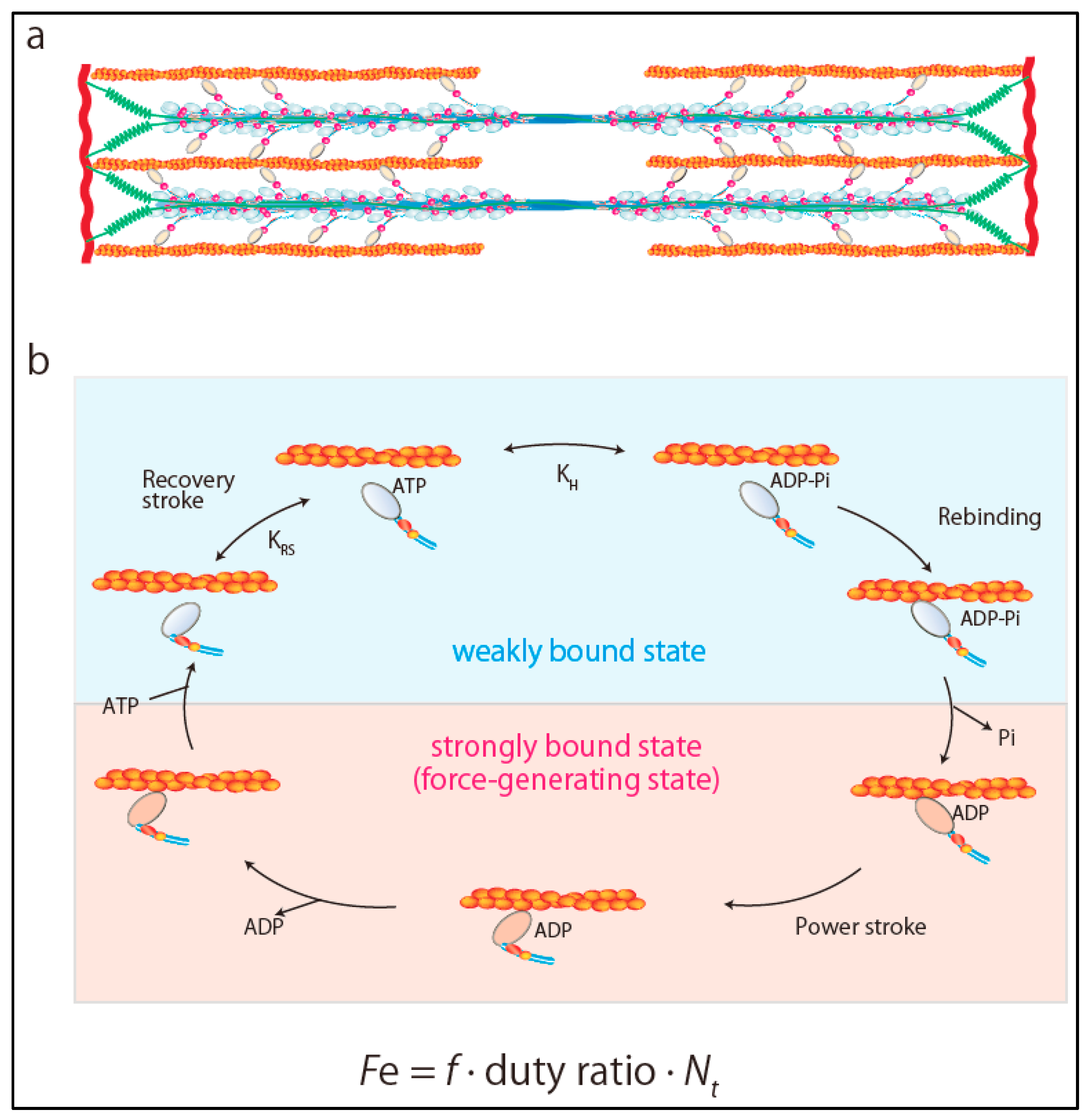
2. Ensemble Force and Sarcomere Power Output
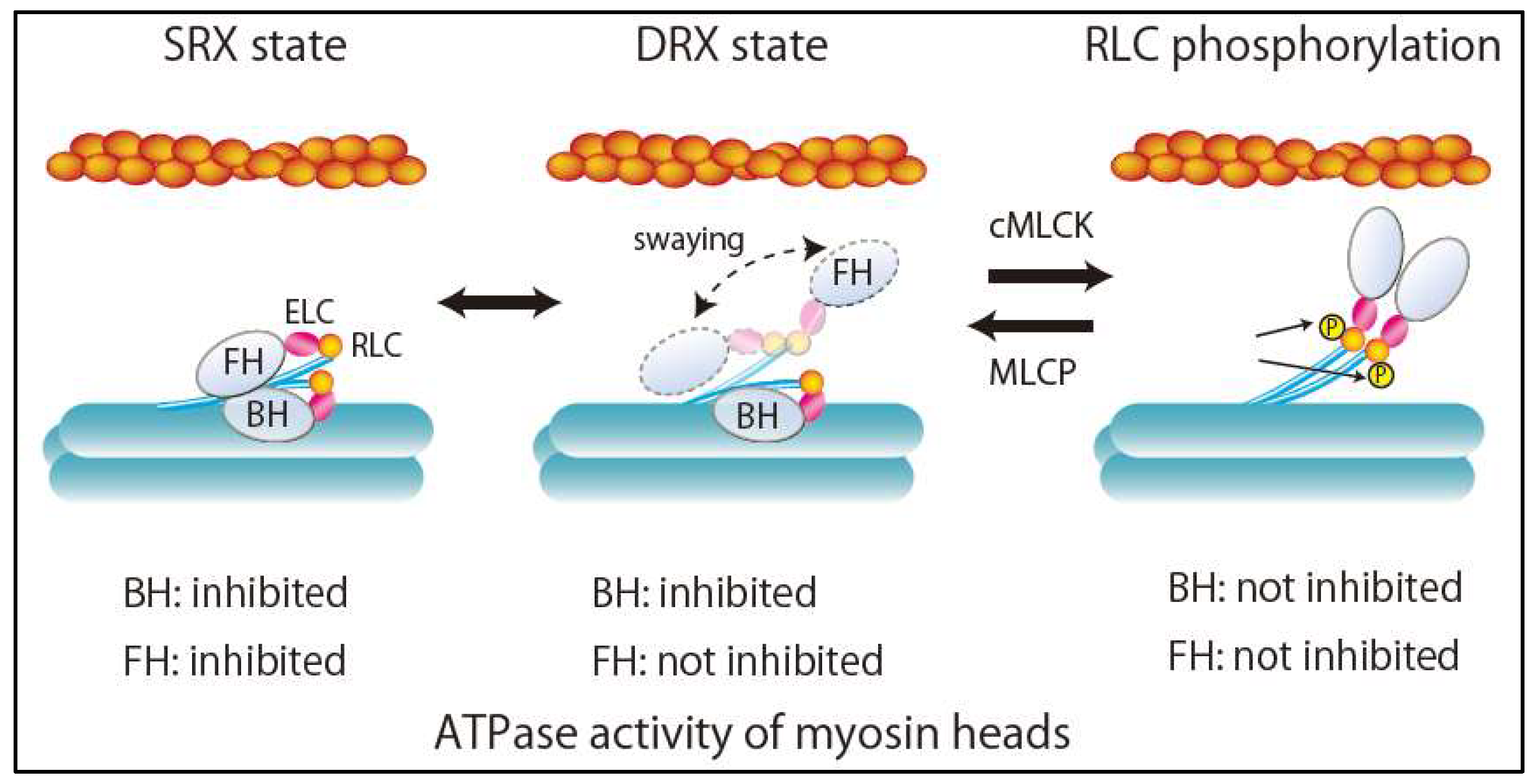
3. Super-Relaxed State in the Heart
4. Hypertrophic Cardiomyopathy (HCM)
4.1. Hypocontractile Hypothesis
4.2. Hypercontractile Hypothesis
4.3. New Drug Therapy Hypertrophic Obstructive Cardiomyopathy (HOCM)
5. Dilated Cardiomyopathy (DCM)
5.1. Genetic Causes of DCM
5.2. Challenges in the Development of New Drug Types for Systolic Heart Failure
5.2.1. Omecamtiv Mecarbil (OM)
5.2.2. Other Target Molecules for Direct Sarcomere Modulators
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| HCM | Hypertrophic cardiomyopathy |
| DCM | Dilated cardiomyopathy |
| HFrEF | Heart failure with reduced ejection fraction |
| DRX | Disordered relaxed |
| SRX | Super-relaxed |
| BH | Blocked head |
| FH | Free head |
| vRLC | Ventricular regulatory light chain |
| cMLCK | Cardiac myosin regulatory light chain kinase |
| MHC | Myosin heavy chain |
| HOCM | Hypertrophic obstructive cardiomyopathy |
| LVOT | Left ventricular outflow tract |
| hiPSC-CM | Human induced pluripotent stem cell derived cardiac myocyte |
| OM | Omecamtiv mecarbil |
References
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [Green Version]
- Spudich, J.A.; Aksel, T.; Bartholomew, S.R.; Nag, S.; Kawana, M.; Yu, E.C.; Sarkar, S.S.; Sung, J.; Sommese, R.F.; Sutton, S.; et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human beta-cardiac myosin. J. Exp. Biol. 2016, 219, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Spudich, J.A. Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 2014, 106, 1236–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houdusse, A.; Sweeney, H.L. How Myosin Generates Force on Actin Filaments. Trends Biochem. Sci. 2016, 41, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommese, R.F.; Sung, J.; Nag, S.; Sutton, S.; Deacon, J.C.; Choe, E.; Leinwand, L.A.; Ruppel, K.; Spudich, J.A. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc. Natl. Acad. Sci. USA 2013, 110, 12607–12612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, A.S.; Kooiker, K.B.; Sarkar, S.S.; Liu, C.; Bernstein, D.; Spudich, J.A.; Ruppel, K.M. Early-Onset Hypertrophic Cardiomyopathy Mutations Significantly Increase the Velocity, Force, and Actin-Activated ATPase Activity of Human beta-Cardiac Myosin. Cell Rep. 2016, 17, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Aksel, T.; Choe Yu, E.; Sutton, S.; Ruppel, K.M.; Spudich, J.A. Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 2015, 11, 910–920. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.W.; Li, A.; Dos Remedios, C.G.; Cooke, R. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys. Rev. 2015, 7, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Alamo, L.; Koubassova, N.; Pinto, A.; Gillilan, R.; Tsaturyan, A.; Padron, R. Lessons from a tarantula: New insights into muscle thick filament and myosin interacting-heads motif structure and function. Biophys. Rev. 2017, 9, 461–480. [Google Scholar] [CrossRef] [Green Version]
- Robert-Paganin, J.; Auguin, D.; Houdusse, A. Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat. Commun. 2018, 9, 4019. [Google Scholar] [CrossRef] [Green Version]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padron, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 2017, 6, e24634. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci. 2018, 19, 2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marian, A.J. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet 2000, 355, 58–60. [Google Scholar] [CrossRef]
- Witjas-Paalberends, E.R.; Piroddi, N.; Stam, K.; van Dijk, S.J.; Oliviera, V.S.; Ferrara, C.; Scellini, B.; Hazebroek, M.; ten Cate, F.J.; van Slegtenhorst, M.; et al. Mutations in MYH7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 99, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Cuda, G.; Fananapazir, L.; Epstein, N.D.; Sellers, J.R. The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 1997, 18, 275–283. [Google Scholar] [CrossRef]
- Birket, M.J.; Ribeiro, M.C.; Kosmidis, G.; Ward, D.; Leitoguinho, A.R.; van de Pol, V.; Dambrot, C.; Devalla, H.D.; Davis, R.P.; Mastroberardino, P.G.; et al. Contractile Defect Caused by Mutation in MYBPC3 Revealed under Conditions Optimized for Human PSC-Cardiomyocyte Function. Cell Rep. 2015, 13, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Karabina, A.; Kazmierczak, K.; Szczesna-Cordary, D.; Moore, J.R. Myosin regulatory light chain phosphorylation enhances cardiac beta-myosin in vitro motility under load. Arch. Biochem. Biophys. 2015, 580, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.C.; Muthu, P.; Kazmierczak, K.; Liang, J.; Huang, W.; Irving, T.C.; Kanashiro-Takeuchi, R.M.; Hare, J.M.; Szczesna-Cordary, D. Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2015, 112, E4138–E4146. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Szczesna-Cordary, D. Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J. Muscle Res. Cell Motil. 2015, 36, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Guhathakurta, P.; Prochniewicz, E.; Roopnarine, O.; Rohde, J.A.; Thomas, D.D. A Cardiomyopathy Mutation in the Myosin Essential Light Chain Alters Actomyosin Structure. Biophys. J. 2017, 113, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.R.; Leinwand, L.; Warshaw, D.M. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res. 2012, 111, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgakopoulos, D.; Christe, M.E.; Giewat, M.; Seidman, C.M.; Seidman, J.G.; Kass, D.A. The pathogenesis of familial hypertrophic cardiomyopathy: Early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation. Nat. Med. 1999, 5, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Tyska, M.J.; Hayes, E.; Giewat, M.; Seidman, C.E.; Seidman, J.G.; Warshaw, D.M. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 2000, 86, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.M.; Wang, Y.; Teekakirikul, P.; Hinson, J.T.; Fatkin, D.; Strouse, S.; Vanburen, P.; Seidman, C.E.; Seidman, J.G.; Maughan, D.W. Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1939–H1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.M.; Fishbaugher, D.E.; Schmitt, J.P.; Wang, Y.; Alpert, N.R.; Seidman, C.E.; Seidman, J.G.; VanBuren, P.; Maughan, D.W. Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H91–H99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belus, A.; Piroddi, N.; Scellini, B.; Tesi, C.; D’Amati, G.; Girolami, F.; Yacoub, M.; Cecchi, F.; Olivotto, I.; Poggesi, C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol. 2008, 586, 3639–3644. [Google Scholar] [CrossRef]
- Keller, D.I.; Coirault, C.; Rau, T.; Cheav, T.; Weyand, M.; Amann, K.; Lecarpentier, Y.; Richard, P.; Eschenhagen, T.; Carrier, L. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. J. Mol. Cell. Cardiol. 2004, 36, 355–362. [Google Scholar] [CrossRef]
- Alpert, N.R.; Mohiddin, S.A.; Tripodi, D.; Jacobson-Hatzell, J.; Vaughn-Whitley, K.; Brosseau, C.; Warshaw, D.M.; Fananapazir, L. Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy-chain mutations. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1097–H1102. [Google Scholar] [CrossRef]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Kawas, R.F.; Anderson, R.L.; Ingle, S.R.B.; Song, Y.H.; Sran, A.S.; Rodriguez, H.M. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J. Biol. Chem. 2017, 292, 16571–16577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, J.A.; Roopnarine, O.; Thomas, D.D.; Muretta, J.M. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. USA 2018, 115, E7486–E7494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, J.A.; Markova, S.; Ueda, Y.; Kim, J.B.; Pascoe, P.J.; Evanchik, M.J.; Green, E.M.; Harris, S.P. A Small Molecule Inhibitor of Sarcomere Contractility Acutely Relieves Left Ventricular Outflow Tract Obstruction in Feline Hypertrophic Cardiomyopathy. PLoS ONE 2016, 11, e0168407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [Green Version]
- Chopra, A.; Kutys, M.L.; Zhang, K.; Polacheck, W.J.; Sheng, C.C.; Luu, R.J.; Eyckmans, J.; Hinson, J.T.; Seidman, J.G.; Seidman, C.E.; et al. Force Generation via beta-Cardiac Myosin, Titin, and alpha-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Dev. Cell 2018, 44, 87–96.e5. [Google Scholar] [CrossRef] [Green Version]
- Dewan, S.; McCabe, K.J.; Regnier, M.; McCulloch, A.D. Insights and Challenges of Multi-Scale Modeling of Sarcomere Mechanics in cTn and Tm DCM Mutants-Genotype to Cellular Phenotype. Front. Physiol. 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Clippinger, S.R.; Cloonan, P.E.; Greenberg, L.; Ernst, M.; Stump, W.T.; Greenberg, M.J. Disrupted mechanobiology links the molecular and cellular phenotypes in familial dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 17831–17840. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Liang, J.; Yuan, C.C.; Kazmierczak, K.; Zhou, Z.; Morales, A.; McBride, K.L.; Fitzgerald-Butt, S.M.; Hershberger, R.E.; Szczesna-Cordary, D. Novel familial dilated cardiomyopathy mutation in MYL2 affects the structure and function of myosin regulatory light chain. FEBS J. 2015, 282, 2379–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Zhou, Z.; Yadav, S.; Gomes, A.V.; Irving, T.C.; Szczesna-Cordary, D. Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2338–E2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, J.P.; Debold, E.P.; Ahmad, F.; Armstrong, A.; Frederico, A.; Conner, D.A.; Mende, U.; Lohse, M.J.; Warshaw, D.; Seidman, C.E.; et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. USA 2006, 103, 14525–14530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T.; et al. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; White, H.D.; Belknap, B.; Winkelmann, D.A.; Forgacs, E. Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine beta-cardiac myosin. Biochemistry 2015, 54, 1963–1975. [Google Scholar] [CrossRef]
- Winkelmann, D.A.; Forgacs, E.; Miller, M.T.; Stock, A.M. Structural basis for drug-induced allosteric changes to human beta-cardiac myosin motor activity. Nat. Commun. 2015, 6, 7974. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.; Solomon, S.D.; Adams, K.F., Jr.; Cleland, J.G.; Ezekowitz, J.A.; Goudev, A.; Macdonald, P.; Metra, M.; et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 2016, 388, 2895–2903. [Google Scholar] [CrossRef] [Green Version]
- Cleland, J.G.; Teerlink, J.R.; Senior, R.; Nifontov, E.M.; Mc Murray, J.J.; Lang, C.C.; Tsyrlin, V.A.; Greenberg, B.H.; Mayet, J.; Francis, D.P.; et al. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: A double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet 2011, 378, 676–683. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Clarke, C.P.; Saikali, K.G.; Lee, J.H.; Chen, M.M.; Escandon, R.D.; Elliott, L.; Bee, R.; Habibzadeh, M.R.; Goldman, J.H.; et al. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: A first-in-man study. Lancet 2011, 378, 667–675. [Google Scholar] [CrossRef]
- Hwang, P.M.; Sykes, B.D. Targeting the sarcomere to correct muscle function. Nat. Rev. Drug Discov. 2015, 14, 313–328. [Google Scholar] [CrossRef]
- Mamidi, R.; Li, J.; Gresham, K.S.; Stelzer, J.E. Cardiac myosin binding protein-C: A novel sarcomeric target for gene therapy. Pflug. Arch. Eur. J. Physiol. 2014, 466, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczesna-Cordary, D.; de Tombe, P.P. Myosin light chain phosphorylation, novel targets to repair a broken heart? Cardiovasc. Res. 2016, 111, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsukamoto, O. Direct Sarcomere Modulators Are Promising New Treatments for Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 226. https://doi.org/10.3390/ijms21010226
Tsukamoto O. Direct Sarcomere Modulators Are Promising New Treatments for Cardiomyopathies. International Journal of Molecular Sciences. 2020; 21(1):226. https://doi.org/10.3390/ijms21010226
Chicago/Turabian StyleTsukamoto, Osamu. 2020. "Direct Sarcomere Modulators Are Promising New Treatments for Cardiomyopathies" International Journal of Molecular Sciences 21, no. 1: 226. https://doi.org/10.3390/ijms21010226
APA StyleTsukamoto, O. (2020). Direct Sarcomere Modulators Are Promising New Treatments for Cardiomyopathies. International Journal of Molecular Sciences, 21(1), 226. https://doi.org/10.3390/ijms21010226