Neutrophil Metabolic Shift during Their Lifecycle: Impact on Their Survival and Activation

Abstract
:1. Introduction
2. Available Metabolites in the Bone Marrow, Plasma, and Inflamed Tissues
2.1. Bone Marrow Homeostasis
2.2. Plasma Homeostasis
2.3. Tissue Homeostasis
2.4. Available Metabolites in the Inflamed Tissue Microenvironment
3. Neutrophil Metabolism
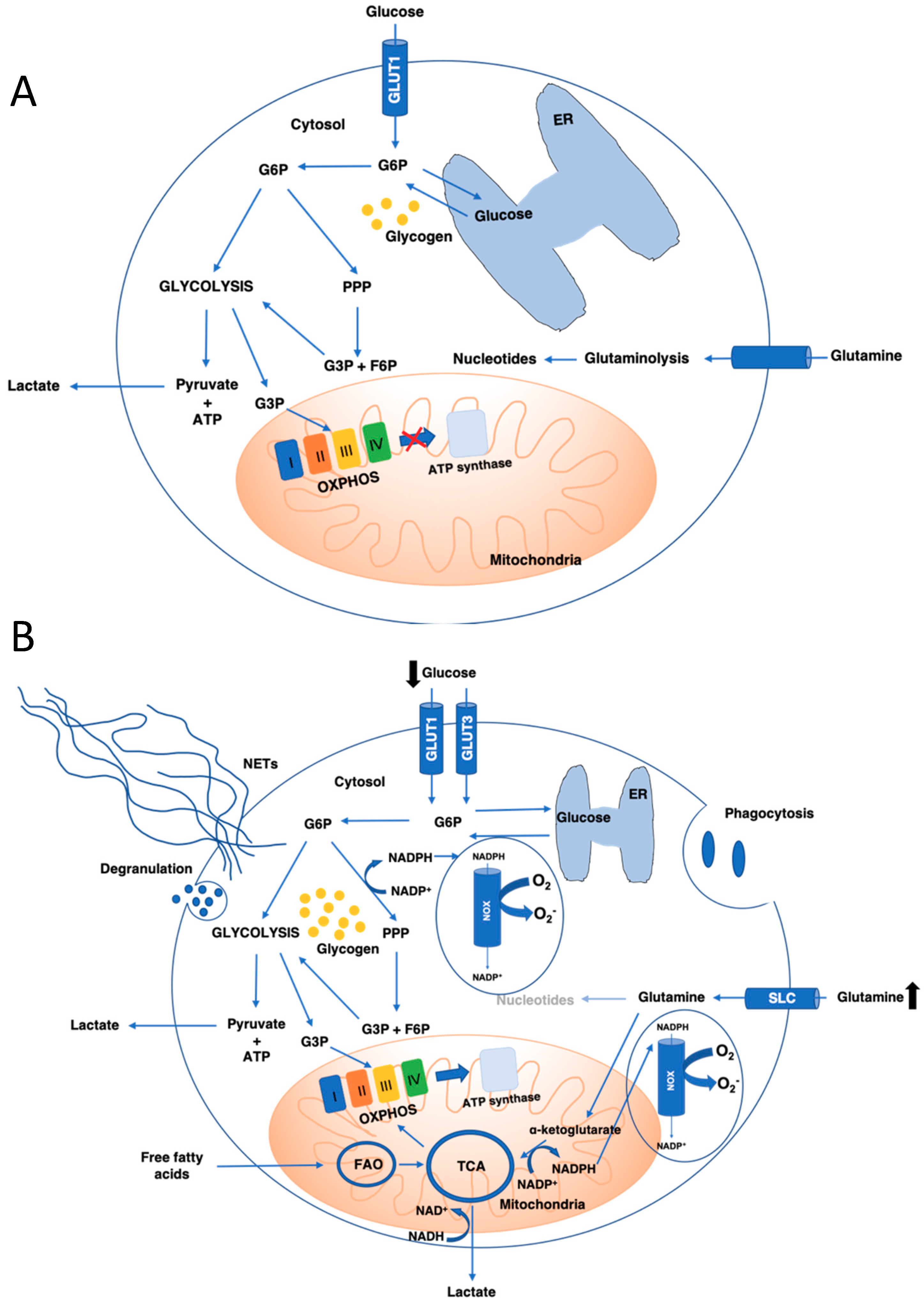
3.1. Glucose Metabolism: Glycolysis and Pentose Phosphate Pathways
3.2. Glutamine Metabolism
3.3. Mitochondrial Metabolism: TCA Cycle, OXPHOS, and Fatty Acid Oxidation
4. Changes in Metabolism
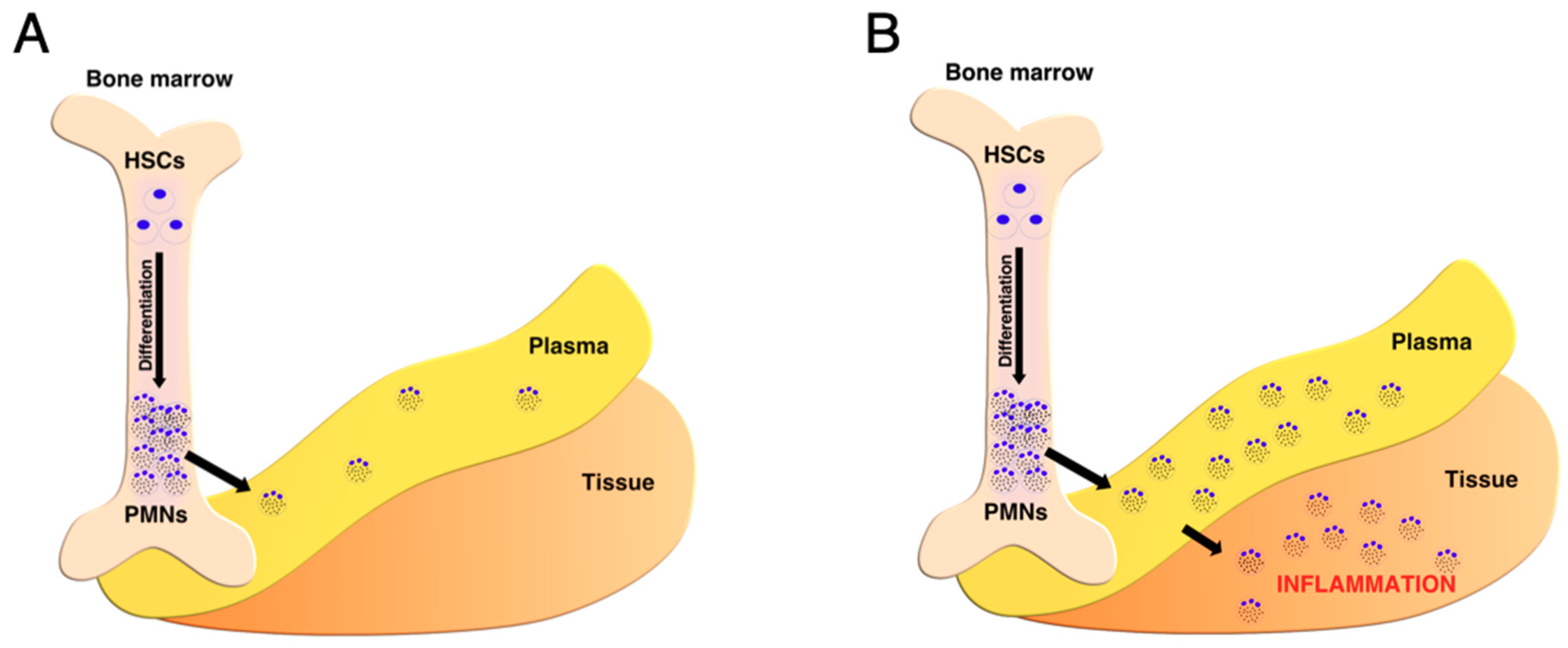
4.1. Metabolic Shift from Hematopoietic Stem Cells to Mature Neutrophils
4.2. Neutrophils’ Metabolic Shift from Plasma to Tissues
4.3. Neutrophils Metabolic Shift upon Neutrophil Antimicrobial Functions Activation
4.4. Metabolic Shift during Infection
5. Importance of Metabolic Shifts in Neutrophil Population Heterogeneity
5.1. Tumor-Associated Neutrophils
5.2. Low-Density Neutrophils
6. Conclusions
Funding
Conflicts of Interest
References
- Maianski, N.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ. 2004, 11, 4401320. [Google Scholar] [CrossRef]
- Mecklenburgh, K.I.; Walmsley, S.R.; Cowburn, A.S.; Wiesener, M.; Reed, B.J.; Upton, P.D.; Deighton, J.; Greening, A.P.; Chilvers, E.R. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood 2002, 100, 3008–3016. [Google Scholar] [CrossRef]
- Walmsley, S.R.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; Chilvers, E.R. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Monceaux, V.; Chiche-Lapierre, C.; Chaput, C.; Witko-Sarsat, V.; Prevost, M.C.; Taylor, C.T.; Ungeheuer, M.N.; Sansonetti, P.J.; Marteyn, B.S. Anoxia and glucose supplementation preserve neutrophil viability and function. Blood 2016, 128, 993–1002. [Google Scholar] [CrossRef]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular Metabolism and Macrophage Functional Polarization. Int. Rev. Immunol. 2014, 34, 82–100. [Google Scholar] [CrossRef]
- Genard, G.; Wera, A.C.; Huart, C.; Le Calve, B.; Penninckx, S.; Fattaccioli, A.; Tabarrant, T.; Demazy, C.; Ninane, N.; Heuskin, A.C.; et al. Proton irradiation orchestrates macrophage reprogramming through NFκB signaling. Cell Death Dis. 2018, 9, 728. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Grecian, R.; Whyte, M.K.; Walmsley, S.R. The role of neutrophils in cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Sadiku, P.; Walmsley, S.R. Hypoxia and the regulation of myeloid cell metabolic imprinting: Consequences for the inflammatory response. EMBO Rep. 2019, 20, e47388. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269. [Google Scholar] [CrossRef] [Green Version]
- Stegenga, M.E.; van der Crabben, S.N.; Blümer, R.M.; Levi, M.; Meijers, J.C.; Serlie, M.J.; Tanck, M.W.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood 2008, 112, 82–89. [Google Scholar] [CrossRef]
- Fainsod-Levi, T.; Gershkovitz, M.; Völs, S.; Kumar, S.; Khawaled, S.; Sagiv, J.Y.; Sionov, R.V.; Grunewald, M.; Keshet, E.; Granot, Z. Hyperglycemia Impairs Neutrophil Mobilization Leading to Enhanced Metastatic Seeding. Cell Rep. 2017, 21, 2384–2392. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, X.; Zhou, X.; Wang, D. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef]
- Thom, S.R.; Bhopale, V.M.; Yu, K.; Huang, W.; Kane, M.A.; Margolis, D.J. Neutrophil microparticle production and inflammasome activation by hyperglycemia due to cytoskeletal instability. J. Biol. Chem. 2017, 292, 18312–18324. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Procopio, J.; Lima, M.; Pithon-Curi, T.; Curi, R. Glutamine and glutamate—Their central role in cell metabolism and function. Cell Biochem. Funct. 2003, 21, 1–9. [Google Scholar] [CrossRef]
- Pittman, R.N. Regulation of Tissue Oxygenation. In Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011; Volume 3, pp. 1–100. [Google Scholar]
- Borregaard, N.; Herlin, T. Energy Metabolism of Human Neutrophils during Phagocytosis. J. Clin. Investig. 1982, 70, 550–557. [Google Scholar] [CrossRef]
- Fossati, G.; Moulding, D.A.; Spiller, D.G.; Moots, R.J.; White, M.R.; Edwards, S.W. The Mitochondrial Network of Human Neutrophils: Role in Chemotaxis, Phagocytosis, Respiratory Burst Activation, and Commitment to Apoptosis. J. Immunol. 2003, 170, 1964–1972. [Google Scholar] [CrossRef]
- Robinson, J.; Karnovsky, M.; Karnovsky, M. Glycogen accumulation in polymorphonuclear leukocytes, and other intracellular alterations that occur during inflammation. J. Cell. Biol. 1982, 95, 933–942. [Google Scholar] [CrossRef]
- Ackerman, A.G. Histochemical differentiation during neutrophil development and maturation. Ann. N. Y. Acad. Sci. 1964, 113, 537–565. [Google Scholar] [CrossRef]
- Sadiku, P.; Willson, J.A.; Dickinson, R.S.; Murphy, F.; Harris, A.J.; Lewis, A.; Sammut, D.; Mirchandani, A.S.; Ryan, E.; Watts, E.R.; et al. Prolyl hydroxylase 2 inactivation enhances glycogen storage and promotes excessive neutrophilic responses. J. Clin. Investig. 2017, 127, 3407–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinnars, E.; Bergstöm, J.; Fürst, P. Influence of the Postoperative State on the Intracellular Free Amino Acids in Human Muscle Tissue. Ann. Surg. 1975, 182, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Häussinger, D.; Soboll, S.; Meijer, A.J.; Gerok, W.; Tager, J.M.; Sies, H. Role of plasma membrane transport in hepatic glutamine metabolism. Eur. J. Biochem. 1985, 152, 597–603. [Google Scholar] [CrossRef]
- Quesada, A.; Medina, M.; Márquez, J.; nchez-Jiménez, F.; de Castro, N.I. Contribution by host tissues to circulating glutamine in mice inoculated with Ehrlich ascites tumor cells. Cancer Res. 1988, 48, 1551–1553. [Google Scholar]
- McKeown, S. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Carreau, A.; Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Winlove, C.; Michel, C. Oxygen Partial Pressure in Outer Layers of Skin of Human Finger Nail Folds. J. Physiol. 2003, 549, 855–863. [Google Scholar] [CrossRef]
- Muller, M.; Padberg, W.; Schindler, E.; Sticher, J.; Osmer, C.; Friemann, S.; Hempelmann, G. Renocortical Tissue Oxygen Pressure Measurements in Patients Undergoing Living Donor Kidney Transplantation. Anesth. Analg. 1998, 87, 474–476. [Google Scholar]
- Bowden, S.D.; Rowley, G.; Hinton, J.C.; Thompson, A. Glucose and Glycolysis Are Required for the Successful Infection of Macrophages and Mice by Salmonella enterica Serovar Typhimurium. Infect. Immun. 2009, 77, 3117–3126. [Google Scholar] [CrossRef] [Green Version]
- Xavier, M.N.; Winter, M.G.; Spees, A.M.; Den Hartigh, A.B.; Nguyen, K.; Roux, C.M.; Silva, T.M.; Atluri, V.L.; Kerrinnes, T.; Keestra, A.M.; et al. PPARγ-Mediated Increase in Glucose Availability Sustains Chronic Brucella abortus Infection in Alternatively Activated Macrophages. Cell Host Microbe 2013, 14, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Huen, S.C.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Kim, H. The Roles of Glutamine in the Intestine and Its Implication in Intestinal Diseases. Int. J. Mol. Sci. 2017, 18, 1051. [Google Scholar] [CrossRef] [Green Version]
- Hulsewé, K.; van der Hulst, R.; van Acker, B.; von Meyenfeldt, M.F.; Soeters, P.B. Inflammation rather than nutritional depletion determines glutamine concentrations and intestinal permeability. Clin. Nutr. 2004, 23, 1209–1216. [Google Scholar]
- Djoko, K.Y.; Phan, M.D.; Peters, K.M.; Walker, M.J.; Schembri, M.A.; McEwan, A.G. Interplay between tolerance mechanisms to copper and acid stress in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, 6818–6823. [Google Scholar]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef]
- Schwartz, R.S.; Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar]
- Arena, E.T.; Tinevez, J.-Y.; Nigro, G.; Sansonetti, P.J.; Marteyn, B.S. The infectious hypoxia: Occurrence and causes during Shigella infection. Microbes Infect. 2017, 19, 157–165. [Google Scholar] [CrossRef]
- Tinevez, J.Y.; Arena, E.T.; Anderson, M.; Nigro, G.; Injarabian, L.; André, A.; Ferrari, M.; Campbell-Valois, F.X.; Devin, A.; Shorte, S.L.; et al. Shigella-mediated oxygen depletion is essential for intestinal mucosa colonization. Nat. Microbiol. 2019, 4, 2001–2009. [Google Scholar] [CrossRef]
- Werth, N.; Beerlage, C.; Rosenberger, C.; Yazdi, A.S.; Edelmann, M.; Amr, A.; Bernhardt, W.; Von Eiff, C.; Becker, K.; Schäfer, A.; et al. Activation of Hypoxia Inducible Factor 1 Is a General Phenomenon in Infections with Human Pathogens. PLoS ONE 2010, 5, e11576. [Google Scholar] [CrossRef]
- Hajdamowicz, N.H.; Hull, R.C.; Foster, S.J.; Condliffe, A.M. The Impact of Hypoxia on the Host-Pathogen Interaction between Neutrophils and Staphylococcus aureus. Int. J. Mol. Sci. 2019, 20, 5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennewein, J.; Matuszak, J.; Walter, S.; Felmy, B.; Gendera, K.; Schatz, V.; Nowottny, M.; Liebsch, G.; Hensel, M.; Hardt, W.D.; et al. Low-oxygen tensions found in Salmonella-infected gut tissue boost Salmonella replication in macrophages by impairing antimicrobial activity and augmenting Salmonella virulence. Cell Microbiol. 2015, 17, 1833–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustad, T.R.; Harrell, M.I.; Liao, R.; Sherman, D.R. The Enduring Hypoxic Response of Mycobacterium tuberculosis. PLoS ONE 2008, 3, e1502. [Google Scholar] [CrossRef] [PubMed]
- Van Sluis, R.; Bhujwalla, Z.M.; Raghunand, N.; Ballesteros, P.; Alvarez, J.; Cerdán, S.; Galons, J.P.; Gillies, R.J. In vivo imaging of extracellular pH using 1H MRSI. Magnet. Reson. Med. 1999, 41, 743–750. [Google Scholar] [CrossRef]
- Bailey, K.; Wojtkowiak, J.; Hashim, I.A.; Gillies, R. Targeting the Metabolic Microenvironment of Tumors. Adv. Pharmacol. 2012, 65, 63–107. [Google Scholar] [PubMed] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharping, N.; Delgoffe, G. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines 2016, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Rivera, S.; Azcón-Bieto, J.; López-Soriano, F.; Miralpeix, M.; Argilés, J. Amino acid metabolism in tumour-bearing mice. Biochem. J. 1988, 249, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.; Simonsen, D.; Tanaka, K.; Tanaka, T. Free amino acids in growing and regressing ascites cell tumors: Host resistance and chemical agents. Cancer Res. 1956, 16, 970–978. [Google Scholar] [PubMed]
- Márquez, J.; Sánchez-Jiménez, F.; Medina, M.; Quesada, A.R.; de Castro, I. Nitrogen metabolism in tumor bearing mice. Arch. Biochem. Biophys. 1989, 268, 667–675. [Google Scholar] [CrossRef]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef]
- Lewis, D.M.; Park, K.M.; Tang, V.; Xu, Y.; Pak, K.; Eisinger-Mathason, T.K.; Simon, M.C.; Gerecht, S. Intratumoral oxygen gradients mediate sarcoma cell invasion. Proc. Natl. Acad. Sci. USA 2016, 113, 9292–9297. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.S.; Lee, Y.M.; Cheung, Y.Y.; McDermott, D.H.; Murphy, P.M.; De Ravin, S.S.; Mansfield, B.C.; Chou, J.Y. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-β–deficient neutrophils in a congenital neutropenia syndrome. Blood 2010, 116, 2783–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.; Weinstein, D.A.; Lee, Y.; Mansfield, B.C.; Chou, J.Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashan, N.; Potashnik, R.; Hagay, Y.; Moses, S. Impaired glucose transport in polymorphonuclear leukocytes in glycogen storage disease Ib. J. Inherit. Metab. Dis. 1987, 10, 234–241. [Google Scholar] [CrossRef]
- Boztug, K.; Appaswamy, G.; Ashikov, A.; Schäffer, A.A.; Salzer, U.; Diestelhorst, J.; Germeshausen, M.; Brandes, G.; Lee-Gossler, J.; Noyan, F.; et al. A Syndrome with Congenital Neutropenia and Mutations in G6PC3. N. Engl. J. Med. 2009, 360, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga-da-Cunha, M.; Chevalier, N.; Stephenne, X.; Defour, J.P.; Paczia, N.; Ferster, A.; Achouri, Y.; Dewulf, J.P.; Linster, C.L.; Bommer, G.T.; et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 201816143. [Google Scholar] [CrossRef] [Green Version]
- Maratou, E.; Dimitriadis, G.; Kollias, A.; Boutati, E.; Lambadiari, V.; Mitrou, P.; Raptis, S.A. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur. J. Clin. Investig. 2007, 37, 282–290. [Google Scholar] [CrossRef]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.S.; Bozza, F.A.; Hanrahan, C.J.; Wang, L.M.; Wu, Q.; Hoffman, J.M.; Zimmerman, G.A.; Morton, K.A. 18F-fluoro-2-deoxyglucose PET informs neutrophil accumulation and activation in lipopolysaccharide-induced acute lung injury. Nucl. Med. Biol. 2017, 48, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-B.; Kindzelskii, A.L.; Petty, H.R. Hexokinase translocation during neutrophil activation, chemotaxis, and phagocytosis: Disruption by cytochalasin D, dexamethasone, and indomethacin. Cell Immunol. 2002, 218, 95–106. [Google Scholar] [CrossRef]
- Van Raam, B.J.; Sluiter, W.; De Wit, E.; Roos, D.; Verhoeven, A.J.; Kuijpers, T.W. Mitochondrial Membrane Potential in Human Neutrophils Is Maintained by Complex III Activity in the Absence of Supercomplex Organisation. PLoS ONE 2008, 3, e2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, E.P.; Rochael, N.C.; Guimarães-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perner, A.; Nielsen, S.; Rask-Madsen, J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. 2003, 29, 642–645. [Google Scholar] [CrossRef]
- Cooper, M.R.; DeChatelet, L.R.; McCall, C.E.; La Via, M.F.; Spurr, C.L.; Baehner, R.L. Complete Deficiency of Leukocyte Glucose-6-Phosphate Dehydrogenase with Defective Bactericidal Activity. J. Clin. Investig. 1972, 51, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P. Why Is L-Glutamine Metabolism Important to Cells of the Immune System in Health, Postinjury, Surgery or Infection? J. Nutr. 2001, 131, 2515S–2522S. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Rabinowitz, S.J. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. eLife 2018, 7, e33246. [Google Scholar] [CrossRef]
- Curi, T.C.; de Melo, M.P.; de Azevedo, R.B.; Curi, R. Glutamine utilisation by rat neutrophils. Biochem. Soc. Trans. 1997, 25, 249S. [Google Scholar] [CrossRef] [Green Version]
- Pithon-Curi, T.; Levada, A.C.; Lopes, L.R.; Doi, S.Q.; Curi, R. Glutamine plays a role in superoxide production and the expression of p47phox, p22phox and gp91phox in rat neutrophils. Clin. Sci. 2002, 103, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.D.; Schroeder, C.C.; Fok, S.A. An investigation of mitochondrial inner membranes by rapid-freeze deep-etch techniques. J. Cell Biol. 1989, 108, 2233–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, E.; Seelert, H.; Reifschneider, N.H.; Krause, F.; Dencher, N.A.; Vonck, J. Architecture of Active Mammalian Respiratory Chain Supercomplexes. J. Biol. Chem. 2006, 281, 15370–15375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schägger, H. Supercomplexes and subcomplexes of mitochondrial oxidative phosphorylation. Biochim. et Biophys. Acta (BBA)-Bioenergetics 2006, 1757, 1066–1072. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Ledderose, C.; Graf, A.F.; Brix, B.; Birsak, T.; Lee, A.; Zhang, J.; Junger, W.G. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J. Cell. Biol. 2015, 210, 1153–1164. [Google Scholar] [CrossRef]
- Zhou, W.; Cao, L.; Jeffries, J.; Zhu, X.; Staiger, C.J.; Deng, Q. Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis. Models Mech. 2018, 11, dmm.033027. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, C.; Agriesti, F.; Scrima, R.; Falzetti, F.; Di Ianni, M.; Capitanio, N. To breathe or not to breathe: The haematopoietic stem/progenitor cells dilemma. Br. J. Pharmacol. 2013, 169, 1652–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantel, C.; Messina-Graham, S.; Broxmeyer, H.E. Upregulation of nascent mitochondrial biogenesis in mouse hematopoietic stem cells parallels upregulation of CD34 and loss of pluripotency: A potential strategy for reducing oxidative risk in stem cells. Cell Cycle 2010, 9, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Ria, R.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilio, A.; Capitanio, N. Characterization of Mitochondrial and Extra-mitochondrial Oxygen Consuming Reactions in Human Hematopoietic Stem Cells Novel Evidence of the Occurrence of Nad(P)H Oxidase Activity. J. Biol. Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Cooper, D.D.; Hayes, S.F.; Spangrude, G.J. Rhodamine-123 Staining in Hematopoietic Stem Cells of Young Mice Indicates Mitochondrial Activation Rather Than Dye Efflux. Blood 1998, 91, 4106–4117. [Google Scholar] [CrossRef]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating Mitochondrial DNA Mutations Drive Premature Hematopoietic Aging Phenotypes Distinct from Physiological Stem Cell Aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial Role in Stemness and Differentiation of Hematopoietic Stem Cells. Stem Cells Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.-Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC–mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantel, C.R.; O’Leary, H.A.; Chitteti, B.R.; Huang, X.; Cooper, S.; Hangoc, G.; Brustovetsky, N.; Srour, E.F.; Lee, M.R.; Messina-Graham, S.; et al. Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell 2015, 161, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Sun, X.; Chen, Y.; Shen, Y.; Jarvis, J.N. RNA sequencing from human neutrophils reveals distinct transcriptional differences associated with chronic inflammatory states. BMC Med. Genom. 2015, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Subrahmanyam, Y.V.; Yamaga, S.; Prashar, Y.; Lee, H.H.; Hoe, N.P.; Kluger, Y.; Gerstein, M.; Goguen, J.D.; Newburger, P.E.; Weissman, S.M. RNA expression patterns change dramatically in human neutrophils exposed to bacteria. Blood 2001, 97, 2457–2468. [Google Scholar] [CrossRef]
- Thompson, A.R.; Elks, P.M.; Marriott, H.M.; Eamsamarng, S.; Higgins, K.R.; Lewis, A.; Williams, L.; Parmar, S.; Shaw, G.; McGrath, E.E.; et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 2014, 123, 366–376. [Google Scholar] [CrossRef]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, C.M. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [Green Version]
- Bozonet, S.M.; Carr, A.C.; Pullar, J.M.; Vissers, M.C. Enhanced Human Neutrophil Vitamin C Status, Chemotaxis and Oxidant Generation Following Dietary Supplementation with Vitamin C-Rich SunGold Kiwifruit. Nutrients 2015, 7, 2574–2588. [Google Scholar] [CrossRef] [Green Version]
- Vissers, M.; Gunningham, S.P.; Morrison, M.J.; Dachs, G.U.; Currie, M.J. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic. Biol. Med. 2007, 42, 765–772. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C. Ascorbate as a Co-Factor for Fe- and 2-Oxoglutarate Dependent Dioxygenases: Physiological Activity in Tumor Growth and Progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.; Santoso, S.; Chavakis, T. Mechanisms of neutrophil transendothelial migration. Front. Biosci. 2009, 14, 1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodfin, A.; Voisin, M.B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.M.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E.; et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 12, 761. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Furnish, M.; Caino, M.C. Altered mitochondrial trafficking as a novel mechanism of cancer metastasis. Cancer Rep. 2019, e1157. [Google Scholar] [CrossRef]
- Ledderose, C.; Liu, K.; Kondo, Y.; Slubowski, C.J.; Dertnig, T.; Denicolo, S.; Arbab, M.; Hubner, J.; Konrad, K.; Kakhari, M.; et al. Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. JCI 2018, 128, 3583–3594. [Google Scholar] [CrossRef] [Green Version]
- Denk, S.; Neher, M.D.; Messerer, D.A.C.; Wiegner, R.; Nilsson, B.; Rittirsch, D.; Nilsson-Ekdahl, K.; Weckbach, S.; Ignatius, A.; Kalbitz, M.; et al. Complement C5a functions as a master Switch for the pH Balance in neutrophils exerting fundamental immunometabolism effects. J. Immunol. 2017, 198, 4846–4854. [Google Scholar]
- Bao, Y.; Ledderose, C.; Seier, T.; Graf, A.F.; Brix, B.; Chong, E.; Junger, W.G. Mitochondria Regulate Neutrophil Activation by Generating ATP for Autocrine Purinergic Signaling. J. Biol. Chem. 2014, 289, 26794–26803. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.C.A.; Hebeba, C.B.; Hastreiter, A.A.; de Oliveira, D.C.; Naoto Makiyama, E.; Farsky, S.H.P.; Borelli, P.; Fock, R.A. Exogenous glutamine impairs neutrophils migration into infections sites elicited by lipopolysaccharide by a multistep mechanism. Amino Acids 2019, 51, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, P.A.; Struhl, K. Nutrient deprivation elicits a transcriptional and translational inflammatory response coupled to decreased protein synthesis. Cell Rep. 2018, 24, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisdorf, D.J.; Craddock, P.R.; Jacob, H.S. Granulocyes utilize different energy sources for movements and phagocytosis. Inflammation 1982, 6, 245–256. [Google Scholar] [CrossRef]
- Rodríguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.; López-Villegas, E.O.; Sanchez-Garcia, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Arampatzioglou, A.; Papazoglou, D.; Konstantinidis, T.; Crysanthopoulou, A.; Mitsios, A.; Angelidou, I.; Maroulakou, I.; Ritis, K.; Skendros, P. Clarithomycin enhances the antibacterial activity and wound healing capacity in type 2 diabetes mellitus by increasing LL-37 load on neutrophil extracellular traps. Front. Immunol. 2018, 9, 2062. [Google Scholar] [CrossRef]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttacao, A.; Avogaro, A. NETosis delays diabetic wound healing in mice and humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Kummer, U.; Zobeley, J.; Brasen, J.C.; Fahmy, R.; Kindzelskii, A.L.; Petty, A.R.; Clark, A.J.; Petty, H.R. Elevated glucose concentrations promote receptor-independent activation of adherent human neutrophils: An experimental and computation approach. Biophys. J. 2007, 92, 2597–2607. [Google Scholar] [CrossRef] [Green Version]
- Insuela, D.; Coutinho, D.; Martins, M.; Ferrero, M.; Carvalho, V. Neutrophil function impariment is a host susceptibility factor to bacterial infection in diabetes. In Cells of the Immune System; InTechOpen: Rijeka, Croatia, 2019. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.R.; Dickinson, R.S.; Murphy, F.; Thomson, J.P.; Marriott, H.M.; Tavares, A.; Willson, J.; Williams, L.; Lewis, A.; Mirchandani, A.; et al. Hypoxia determines survival outcomes of bacterial infection through HIF-1a-dependent reprogramming of leukocyte metabolism. Sci. Immunol. 2017, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, C.N.; Breda, L.C.; Khan, M.A.; Almeida, S.R.; Camara, N.O.S.; Sweezy, N.; Palaniyar, N. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: A matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front. Immunol. 2017, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Philip, L.M.; Cheung, G.; Vadakepeedika, S.; Hartmut, G.; Sweezy, N.; Palaniyar, N. Regulating NETosis: Increasing pH promotes NADPH oxidase-dependent NETosis. Front. Med. 2018, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Halperin, M.L.; Connors, H.P.; Relman, A.S.; Karnovsky, M.L. Factors that control the effect of pH on glycolysis in leukocytes. J. Biol. Chem. 1969, 244, 384–390. [Google Scholar] [PubMed]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Colgan, S.P. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272. [Google Scholar] [CrossRef]
- Rupp, J.; Gieffers, J.; Klinger, M.; Van Zandbergen, G.; Wrase, R.; Maass, M.; Solbach, W.; Deiwick, J.; Hellwig-Burgel, T. Chlamydia pneumoniae directly interferes with HIF-1α stabilization in human host cells. Cell Microbiol. 2007, 9, 2181–2191. [Google Scholar] [CrossRef]
- Mohapatra, N.P.; Soni, S.; Rajaram, M.V.; Strandberg, K.L.; Gunn, J.S. Type A Francisella tularensis Acid Phosphatases Contribute to Pathogenesis. PLoS ONE 2013, 8, e56834. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.; Samuel, J. Coxiella burnetii Acid Phosphatase Inhibits the Release of Reactive Oxygen Intermediates in Polymorphonuclear Leukocytes. Infect. Immun. 2011, 79, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Stolper, D.A.; Revsbech, N.; Canfield, D.E. Aerobic growth at nanomolar oxygen concentrations. Proc. Natl. Acad. Sci. USA 2010, 107, 18755–18760. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, J.; Shaul, M.E.; Mishalian, I.; Hovav, A.H.; Levy, L.; Zolotriov, L.; Granot, Z.; Fridlender, Z.G. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 2017, 6, e1356965. [Google Scholar] [CrossRef]
- Richardson, J.; Hendrickse, C.; Gao-Smith, F.; Thickett, D. Neutrophil Extracellular Trap Production in Patients with Colorectal Cancer in Vitro. Int. J. Inflamm. 2017, 2017, 4915062. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuvers, M.E.; Muskens, F.; Bezemer, K.; Lambers, M.; Dingemans, A.M.; Groen, H.J.; Smit, E.F.; Hoogsteden, H.C.; Hegmans, J.P.; Aerts, J.G. Arginase-1 mRNA expression correlates with myeloid-derived suppressor cell levels in peripheral blood of NSCLC patients. Lung Cancer 2013, 81, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Wang, Y.M.; Wang, C.L.; Feng, P.H.; Ko, H.W.; Liu, Y.H.; Wu, Y.C.; Chu, Y.; Chung, F.T.; Kuo, C.H.; et al. Population alterations of l-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J. Cancer Res. Clin. 2010, 136, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; nivas Nagaraj Collazo, M.; Gabrilovich, D.I. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, W.; Zhao, T.; Yan, C.; Du, H. Rab7 GTPase controls lipid metabolic signaling in myeloid-derived suppressor cells. Oncotarget 2017, 8, 30123. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Parekh, A.; Das, S.; Parida, S.; Das, C.K.; Dutta, D.; Mallick, S.K.; Wu, P.H.; Kumar, B.P.; Bharti, R.; Dey, G.; et al. Multi-nucleated cells use ROS to induce breast cancer chemo-resistance in vitro and in vivo. Oncogene 2018, 37, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, J.; Ma, S.; Dong, R.; Meng, W.; Ying, M.; Weng, Q.; Chen, Z.; Ma, J.; Fang, Q.; et al. Tumor hypoxia enhances non-small cell lung cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget 2014, 5, 9664–9677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Kim, S.; Hong, B.J.; Lee, C.J.; Kim, Y.E.; Bok, S.; Oh, J.M.; Gwak, S.H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S.; et al. Transcriptomic Analysis Comparing Tumor-Associated Neutrophils with Granulocytic Myeloid-Derived Suppressor Cells and Normal Neutrophils. PLoS ONE 2012, 7, e31524. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, B.E.; Tabariès, S.; Johnson, R.M.; Andrzejewski, S.; Senecal, J.; Lehuédé, C.; Annis, M.G.; Ma, E.H.; Völs, S.; Ramsay, L.; et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility that Facilitates Breast Cancer Liver Metastasis. Cell Rep. 2019, 27, 3902–3915. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Dikshit, M. Metabolic insight in neutrophils in health and disease. Front. Immunol. 2019, 10, 2099. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Physiological Conditions | |||
| Compartment | Oxygen [c] | Glucose [c] | Glutamine [c] |
| Bone marrow | 1.3–2.9% | ? | ? |
| Plasma | 0.9% | 5 mM | 0.5 mM |
| Tissue | 1–11% | ? | 2–20 mM |
| Pathophysiological Conditions | |||
| Compartment | Oxygen [c] | Glucose [c] | Glutamine [c] |
| Bone marrow | ? | ? | ? |
| Plasma | ? | ? | ↓ |
| Tissue | ↓ | ↓ | ↓ |
| Neutrophil Function | Metabolic Requirements | References |
|---|---|---|
| Phagocytosis | Glycolysis | [18] |
| ROS production (NOX) | PPP, Glutaminolysis | [63,72] |
| Degranulation | Glycolysis | [12,110] |
| NET formation | PPP, Glycolysis | [14,63] |
| Chemotaxis/migration | Glycolysis, mitochondrial metabolism | [76,77,111] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Injarabian, L.; Devin, A.; Ransac, S.; Marteyn, B.S. Neutrophil Metabolic Shift during Their Lifecycle: Impact on Their Survival and Activation. Int. J. Mol. Sci. 2020, 21, 287. https://doi.org/10.3390/ijms21010287
Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil Metabolic Shift during Their Lifecycle: Impact on Their Survival and Activation. International Journal of Molecular Sciences. 2020; 21(1):287. https://doi.org/10.3390/ijms21010287
Chicago/Turabian StyleInjarabian, Louise, Anne Devin, Stéphane Ransac, and Benoit S. Marteyn. 2020. "Neutrophil Metabolic Shift during Their Lifecycle: Impact on Their Survival and Activation" International Journal of Molecular Sciences 21, no. 1: 287. https://doi.org/10.3390/ijms21010287
APA StyleInjarabian, L., Devin, A., Ransac, S., & Marteyn, B. S. (2020). Neutrophil Metabolic Shift during Their Lifecycle: Impact on Their Survival and Activation. International Journal of Molecular Sciences, 21(1), 287. https://doi.org/10.3390/ijms21010287