Docker4Circ: A Framework for the Reproducible Characterization of circRNAs from RNA-Seq Data

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
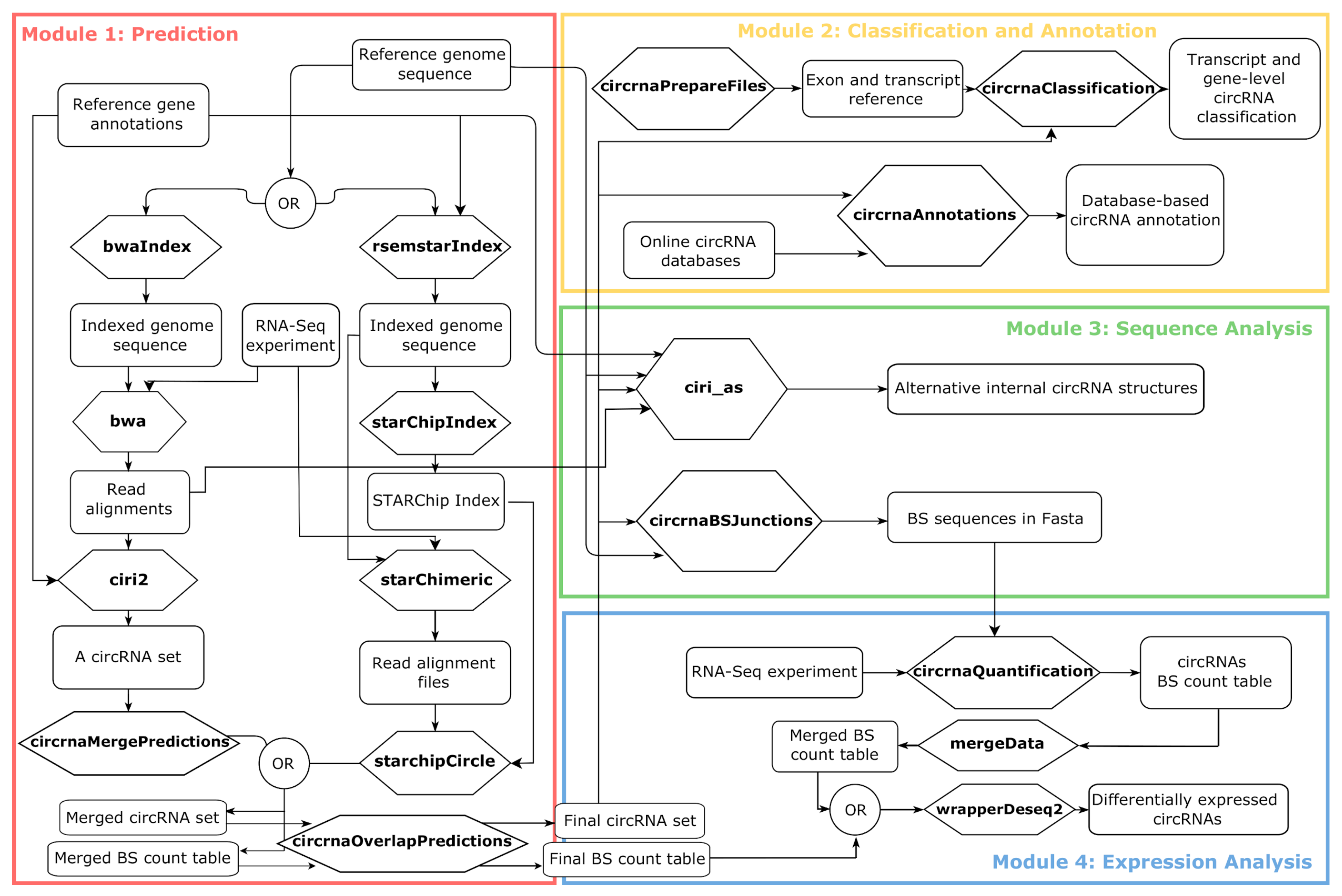
2.1. A Framework to Create Modular Workflows for Reproducible Analysis of circRNA Data
2.1.1. Module 1: circRNAs Prediction
2.1.2. Module 2: circRNAs Classification and Annotation
2.1.3. Module 3: circRNAs Sequence Analysis
2.1.4. Module 4: circRNAs Expression Analysis
2.1.5. How to Integrate a New Functionality in the Framework
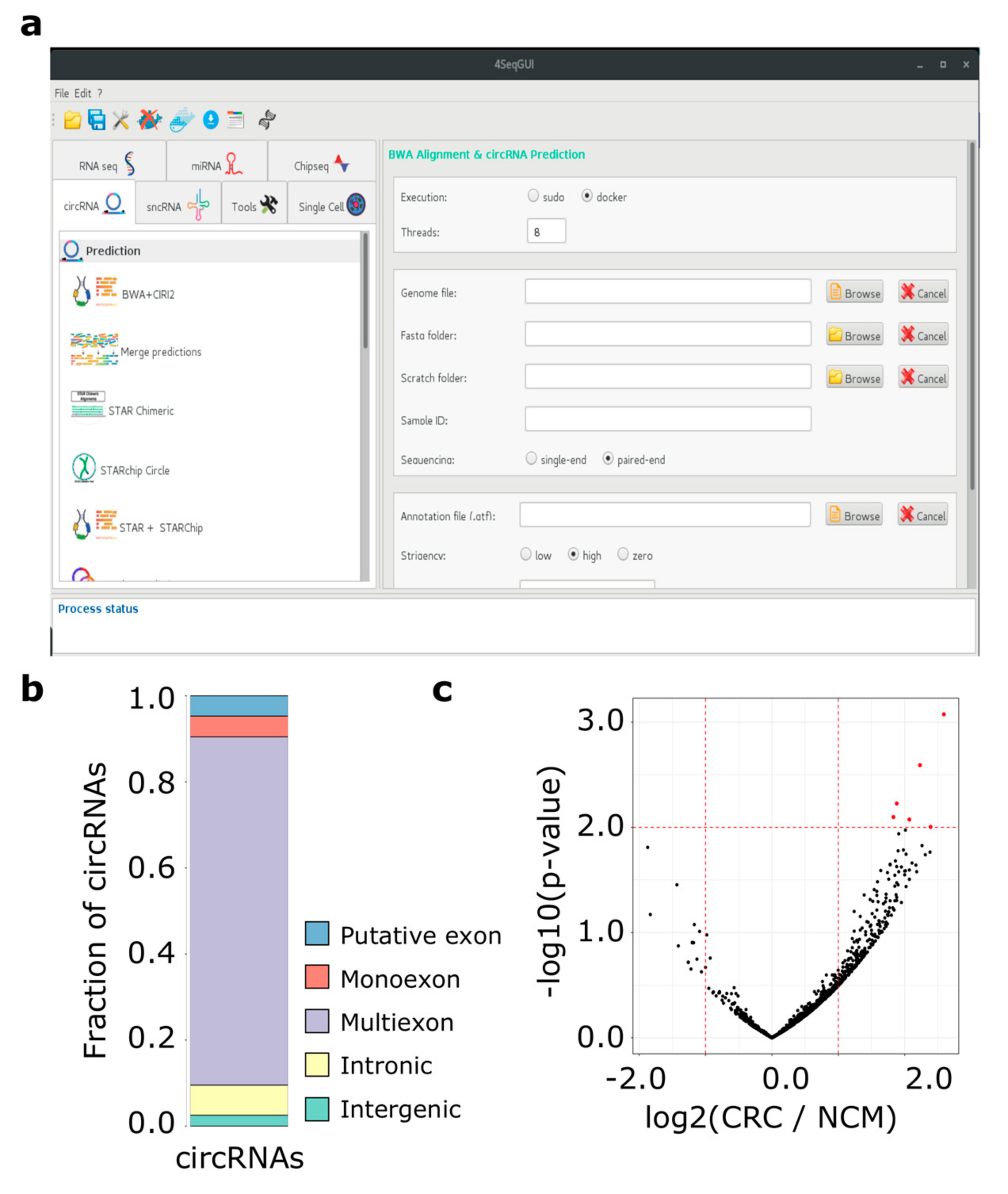
2.2. Examples of the Application of Docker4Circ Framework
2.2.1. Docker4Circ for the Reproducible Analysis of circRNAs Expressed in Colorectal Cancer Cell Lines
2.2.2. Application of Docker4Circ to Directly Quantify circRNAs Expression from CRC Tissue RNA-Seq Data
3. Discussion
4. Materials and Methods
4.1. CircRNAs Prediction
4.2. CircRNAs Classification and Annotations
4.3. CircRNAs Sequence Analysis
4.4. Quantification of circRNAs in RNA-Seq Datasets
4.5. Availability of Source Code and Requirements
4.6. Docker4Circ Running Time Estimation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| circRNA | circular RNAs |
| CRC | colorectal cancer |
| NCM | normal colonic mucosa |
| AS | alternative splicing |
| BS | back-splicing |
References
- Salzman, J. Circular RNA Expression: Its Potential Regulation and Function. Trends Genet. 2016, 32, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Szabo, L.; Salzman, J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, T.; Xiao, J. Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine 2018, 34, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Zhang, L.; Zheng, M.; Sun, X.; Lu, Y.; Liu, P. Circ2Disease: A manually curated database of experimentally validated circRNAs in human disease. Sci. Rep. 2018, 8, 11018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, Y.; Chen, B.; Zhao, J.; Yu, S.; Tang, Y.; Zheng, Q.; Li, Y.; Wang, P.; He, X.; et al. exoRBase: A database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018, 46, D106–D112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Feng, J.; Chen, K.; Ma, Y.; Gong, J.; Cai, F.; Jin, Y.; Gao, Y.; Xia, L.; Chang, H.; et al. CSCD: A database for cancer-specific circular RNAs. Nucleic Acids Res. 2018, 46, D925–D929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019, baz003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhao, F. Computational Strategies for Exploring Circular RNAs. Trends Genet. 2018, 34, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Metge, F.; Czaja-Hasse, L.F.; Reinhardt, R.; Dieterich, C. FUCHS-towards full circular RNA characterization using RNAseq. PeerJ 2017, 5, e2934. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wang, J.; Zheng, Y.; Zhang, J.; Chen, S.; Zhao, F. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat. Commun. 2016, 7, 12060. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xie, X.; Zhou, J.; Sheng, M.; Yin, X.; Ko, E.A.; Zhou, T.; Gu, W. Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics 2017, 33, 2131–2139. [Google Scholar] [CrossRef]
- Cheng, J.; Metge, F.; Dieterich, C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 2016, 32, 1094–1096. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Xiang, Y.; Xia, S.; Liu, H.; Wang, J.; Ozguc, F.M.; Lei, L.; Kong, R.; Diao, L.; He, C.; et al. CircView: A visualization and exploration tool for circular RNAs. Brief. Bioinform. 2018, 19, 1310–1316. [Google Scholar] [CrossRef]
- Coscujuela Tarrero, L.; Ferrero, G.; Miano, V.; De Intinis, C.; Ricci, L.; Arigoni, M.; Riccardo, F.; Annaratone, L.; Castellano, I.; Calogero, R.A.; et al. Luminal breast cancer-specific circular RNAs uncovered by a novel tool for data analysis. Oncotarget 2018, 9, 14580–14596. [Google Scholar]
- Gaffo, E.; Bonizzato, A.; Kronnie, G.T.; Bortoluzzi, S. CirComPara: A Multi-Method Comparative Bioinformatics Pipeline to Detect and Study circRNAs from RNA-seq Data. Noncoding RNA 2017, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, D.T.; Fossat, N.; Tam, P.P.L.; Ho, J.W.K. Ularcirc: Visualisation and enhanced analysis of circular RNAs via back and canonical forward splicing. Nucleic Acids Res. 2019, 47, e123. [Google Scholar] [CrossRef] [PubMed]
- Jakobi, T.; Uvarovskii, A.; Dieterich, C. circtools—A one-stop software solution for circular RNA research. Bioinformatics 2018, 35, 2326–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandve, G.K.; Nekrutenko, A.; Taylor, J.; Hovig, E. Ten simple rules for reproducible computational research. PLoS Comput. Biol. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, N.; Alessandrì, L.; Panero, R.; Arigoni, M.; Olivero, M.; Ferrero, G.; Cordero, F.; Beccuti, M.; Calogero, R.A. Reproducible bioinformatics project: A community for reproducible bioinformatics analysis pipelines. BMC Bioinform. 2018, 19, 349. [Google Scholar] [CrossRef] [PubMed]
- Beccuti, M.; Cordero, F.; Arigoni, M.; Panero, R.; Amparore, E.G.; Donatelli, S.; Calogero, R.A. SeqBox: RNAseq/ChIPseq reproducible analysis on a consumer game computer. Bioinformatics 2018, 34, 871–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Akers, N.K.; Schadt, E.E.; Losic, B. STAR Chimeric Post for rapid detection of circular RNA and fusion transcripts. Bioinformatics 2018, 34, 2364–2370. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Conrad, T.O. Acfs: Accurate circRNA identification and quantification from RNA-Seq data. Sci. Rep. 2016, 6, 38820. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Zhang, N.; Han, P.; Moon, B.S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Lee, W.P.; Garrison, E.P.; Marth, G.T. SSW library: An SIMD Smith-Waterman C/C++ library for use in genomic applications. PLoS ONE 2013, 8, e82138. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zhang, X.; Chu, Q.; Lu, S.; Zhou, L.; Lu, X.; Liu, C.; Mao, L.; Ye, C.; Timko, M.P.; et al. The Circular RNA Profiles of Colorectal Tumor Metastatic Cells. Front. Genet. 2018, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Yu, P.; Lin, W.; Okugawa, Y.; Boland, C.R.; Goel, A. A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci. Rep. 2018, 8, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, P.; Wu, W.; Chen, S.; Zheng, Y.; Zhou, L.; Zhang, J.; Cheng, H.; Yan, J.; Zhang, S.; Yang, P.; et al. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Rep. 2019, 26, 3444.e5–3460.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Dataset ID | Reads | circRNAs | AS Events |
|---|---|---|---|
| NCM460_R1 | 66,144,999 | 14,003 | 1,482 |
| NCM460_R2 | 70,945,094 | 16,006 | 1,790 |
| NCM460_R3 | 73,804,226 | 12,413 | 1,078 |
| SW480_R1 | 88,915,933 | 8,627 | 532 |
| SW480_R2 | 97,303,573 | 5,688 | 335 |
| SW480_R3 | 66,144,999 | 7,154 | 470 |
| SW620_R1 | 91,406,400 | 1,0216 | 790 |
| SW620_R2 | 67,013,355 | 4,624 | 214 |
| SW620_R3 | 69,789,394 | 6,541 | 332 |
| Average | 76,829,774.78 | 9,474.67 | 780.33 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrero, G.; Licheri, N.; Coscujuela Tarrero, L.; De Intinis, C.; Miano, V.; Calogero, R.A.; Cordero, F.; De Bortoli, M.; Beccuti, M. Docker4Circ: A Framework for the Reproducible Characterization of circRNAs from RNA-Seq Data. Int. J. Mol. Sci. 2020, 21, 293. https://doi.org/10.3390/ijms21010293
Ferrero G, Licheri N, Coscujuela Tarrero L, De Intinis C, Miano V, Calogero RA, Cordero F, De Bortoli M, Beccuti M. Docker4Circ: A Framework for the Reproducible Characterization of circRNAs from RNA-Seq Data. International Journal of Molecular Sciences. 2020; 21(1):293. https://doi.org/10.3390/ijms21010293
Chicago/Turabian StyleFerrero, Giulio, Nicola Licheri, Lucia Coscujuela Tarrero, Carlo De Intinis, Valentina Miano, Raffaele Adolfo Calogero, Francesca Cordero, Michele De Bortoli, and Marco Beccuti. 2020. "Docker4Circ: A Framework for the Reproducible Characterization of circRNAs from RNA-Seq Data" International Journal of Molecular Sciences 21, no. 1: 293. https://doi.org/10.3390/ijms21010293
APA StyleFerrero, G., Licheri, N., Coscujuela Tarrero, L., De Intinis, C., Miano, V., Calogero, R. A., Cordero, F., De Bortoli, M., & Beccuti, M. (2020). Docker4Circ: A Framework for the Reproducible Characterization of circRNAs from RNA-Seq Data. International Journal of Molecular Sciences, 21(1), 293. https://doi.org/10.3390/ijms21010293