In Situ Proteolysis Condition-Induced Crystallization of the XcpVWX Complex in Different Lattices



Abstract
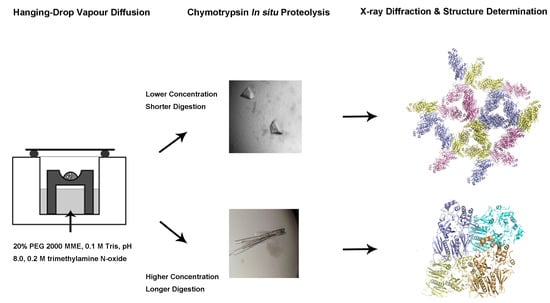
:
1. Introduction
2. Results
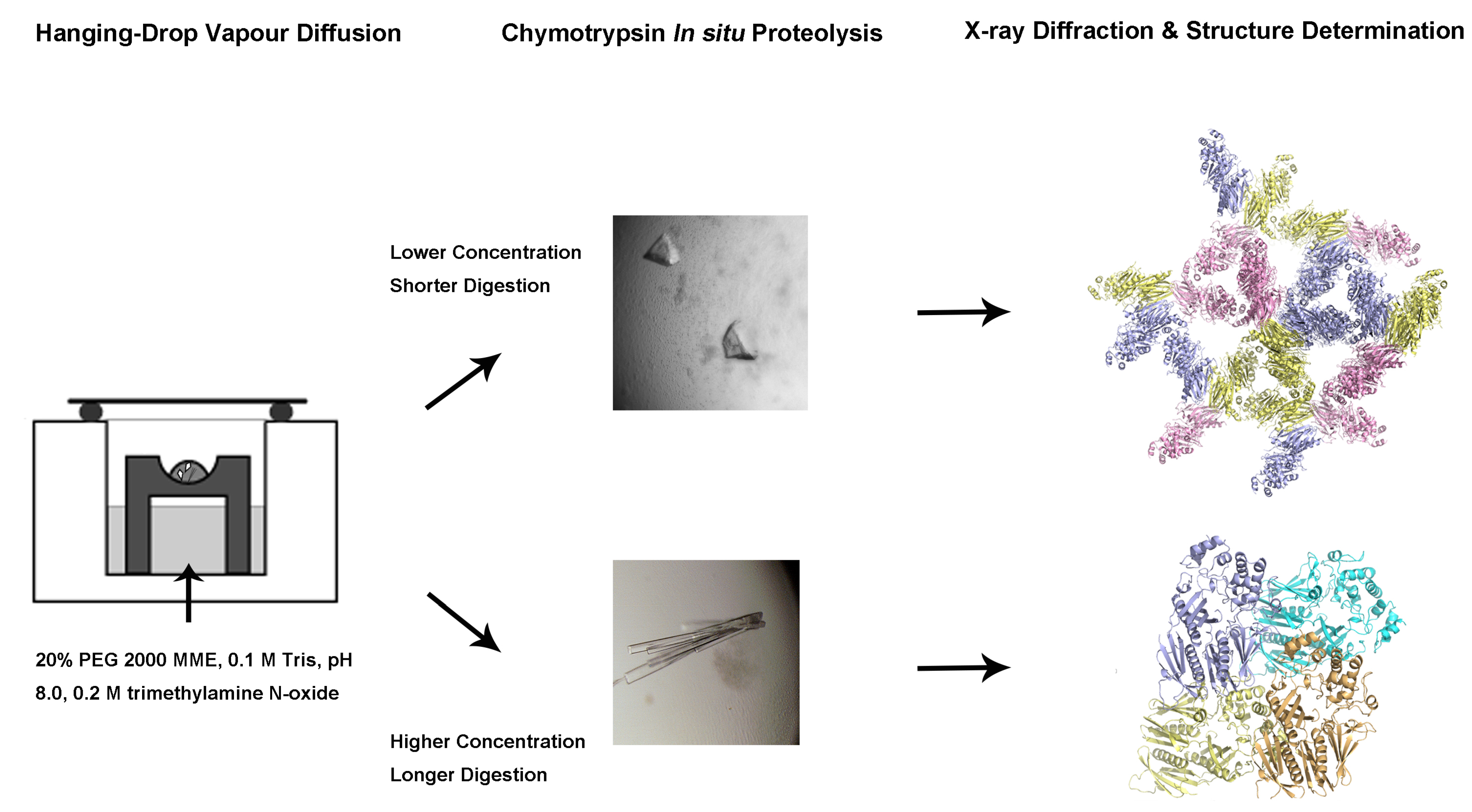
2.1. Purification of the XcpVWX Complex
2.2. Changes In In Situ Proteolysis Conditions Have Led to Distinct XcpVWX Complex Crystal Forms
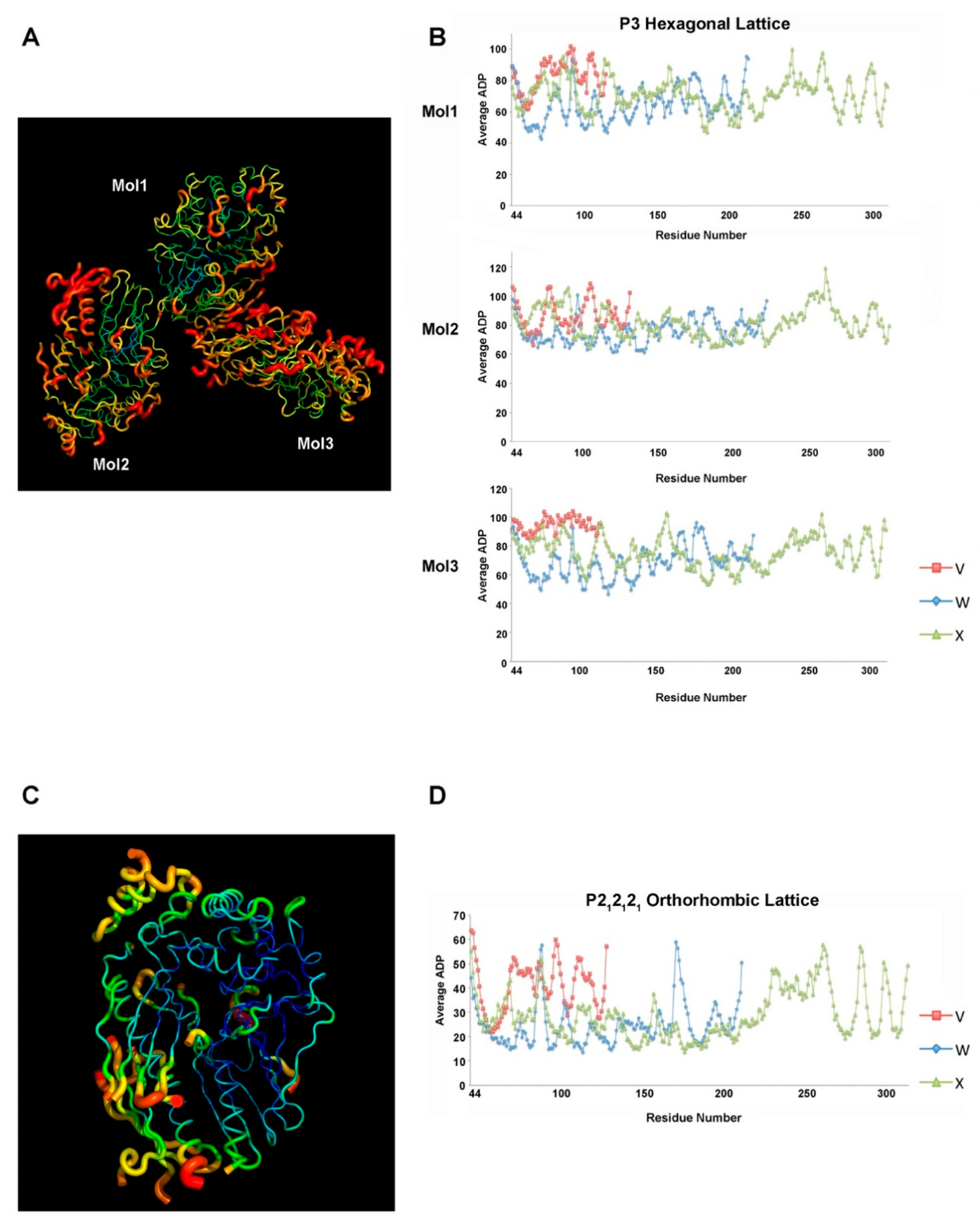
2.3. Different Crystal Forms of the XcpVWX Complex Demonstrate Different Crystal Lattices
2.4. Atom Displacement Parameter Analysis Reveals a Different Flexibility of Molecules in the Two Lattices
3. Discussion
4. Materials and Methods
4.1. Cloning of the Soluble Form of Minor Pseudopilins
4.2. Expression and Purification of Xcp-V, -W, and -X, and the XcpVWX Complex
4.3. Crystallization and Structure Determination
4.4. Calculation of ADP
4.5. Calculation of the Inter-Molecule Contact Formation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Atomic displacement parameter |
| ASU | Asymmetric unit |
| BAVERAGE | Averages B Factor over Main and Side Chain Atoms |
| CT | Chymotrypsin |
| IPTG | Isopropyl β- d-1-thiogalactopyranoside |
| LB | Luria-Bertani Broth |
| MME | Monomethyl Ether |
| NTA | nitrilotriacetic acid |
| PDB | Protein Data Bank |
| PEG | Polyethene glycol |
| PHENIX | Python-based Hierarchical Environment for Integrated Xtallography |
| TEV | Tobacco Etch Virus |
| TMAO | Trimethylamine N-oxide |
| Trx | Thioredoxin |
References
- Smyth, M.S.; Martin, J.H. X ray crystallography. Mol. Pathol. 2000, 53, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-ray crystallography over the past decade for novel drug discovery—Where are we heading next? Expert Opin. Drug Discov. 2015, 10, 975–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srajer, V.; Schmidt, M. Watching Proteins Function with Time-resolved X-ray Crystallography. J. Phys. D Appl. Phys. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Kohn, J.E.; Afonine, P.V.; Ruscio, J.Z.; Adams, P.D.; Head-Gordon, T. Evidence of functional protein dynamics from X-ray crystallographic ensembles. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef]
- Moraes, I.; Evans, G.; Sanchez-Weatherby, J.; Newstead, S.; Stewart, P.D. Membrane protein structure determination—The next generation. Biochim. Biophys. Acta 2014, 1838, 78–87. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Oeffner, R.D.; Wrobel, A.G.; Ojala, J.R.; Tryggvason, K.; Lohkamp, B.; Read, R.J. Ab initio solution of macromolecular crystal structures without direct methods. Proc. Natl. Acad. Sci. USA 2017, 114, 3637–3641. [Google Scholar] [CrossRef] [Green Version]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008, 275, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Russo Krauss, I.; Merlino, A.; Vergara, A.; Sica, F. An overview of biological macromolecule crystallization. Int. J. Mol. Sci. 2013, 14, 11643–11691. [Google Scholar] [CrossRef] [Green Version]
- Chayen, N.E.; Saridakis, E. Protein crystallization: From purified protein to diffraction-quality crystal. Nat. Methods 2008, 5, 147–153. [Google Scholar] [CrossRef]
- Acharya, K.R.; Lloyd, M.D. The advantages and limitations of protein crystal structures. Trends Pharmacol. Sci. 2005, 26, 10–14. [Google Scholar] [CrossRef]
- Dauter, Z.; Wlodawer, A. Progress in protein crystallography. Protein Pept. Lett. 2016, 23, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, J.; Spellmon, N.; Zhang, Y.; Doughan, M.; Li, C.; Yang, Z. Protein crystallization: Eluding the bottleneck of X-ray crystallography. AIMS Biophys. 2017, 4, 557–575. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.; Joachimiak, A.; Otwinowski, Z.; Minor, W. Structural genomics: Keeping up with expanding knowledge of the protein universe. Curr. Opin. Struct. Biol. 2007, 17, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Toole, N.; Grabowski, M.; Otwinowski, Z.; Minor, W.; Cygler, M. The structural genomics experimental pipeline: Insights from global target lists. Proteins 2004, 56, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Bain, K.; Bonanno, J.B.; Buchanan, M.; Henderson, D.; Lorimer, D.; Marsh, C.; Reynes, J.A.; Sauder, J.M.; Schwinn, K.; et al. High-throughput limited proteolysis/mass spectrometry for protein domain elucidation. J. Struct. Funct. Genom. 2005, 6, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Deisenhofer, J.; Colman, P.M.; Matsushima, M.; Palm, W. Crystallographic structure studies of an IgG molecule and an Fc fragment. Nature 1976, 264, 415–420. [Google Scholar] [CrossRef]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72–95. [Google Scholar] [CrossRef] [Green Version]
- Derewenda, Z.S. Application of protein engineering to enhance crystallizability and improve crystal properties. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Derewenda, Z.S. Rational protein crystallization by mutational surface engineering. Structure 2004, 12, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.A.; Muzzin, O.; Chlenov, M.; Sun, J.L.; Olson, C.A.; Weinman, O.; Trester-Zedlitz, M.L.; Darst, S.A. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol. Cell 2002, 9, 527–539. [Google Scholar] [CrossRef]
- Bochkarev, A.; Barwell, J.A.; Pfuetzner, R.A.; Bochkareva, E.; Frappier, L.; Edwards, A.M. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 1996, 84, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Wernimont, A.; Edwards, A. In situ proteolysis to generate crystals for structure determination: An update. PLoS ONE 2009, 4, e5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, A.; Xu, X.; Edwards, A.M.; Midwest Center for Structural, G.; Structural Genomics, C.; Chang, C.; Chruszcz, M.; Cuff, M.; Cymborowski, M.; Di Leo, R.; et al. In situ proteolysis for protein crystallization and structure determination. Nat Methods 2007, 4, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Auperin, T.C.; Tong, L. The use of in situ proteolysis in the crystallization of murine CstF-77. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 135–138. [Google Scholar] [CrossRef]
- Mandel, C.R.; Gebauer, D.; Zhang, H.; Tong, L. A serendipitous discovery that in situ proteolysis is essential for the crystallization of yeast CPSF-100 (Ydh1p). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 1041–1045. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Roversi, P.; Espina, M.; Deane, J.E.; Birket, S.; Picking, W.D.; Blocker, A.; Picking, W.L.; Lea, S.M. Expression, limited proteolysis and preliminary crystallographic analysis of IpaD, a component of the Shigella flexneri type III secretion system. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 865–868. [Google Scholar] [CrossRef]
- Derewenda, Z.S.; Vekilov, P.G. Entropy and surface engineering in protein crystallization. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 116–124. [Google Scholar] [CrossRef]
- Moon, A.F.; Mueller, G.A.; Zhong, X.; Pedersen, L.C. A synergistic approach to protein crystallization: Combination of a fixed-arm carrier with surface entropy reduction. Protein Sci. Publ. Protein Soc. 2010, 19, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Appel, W. Chymotrypsin: Molecular and catalytic properties. Clin. Biochem. 1986, 19, 317–322. [Google Scholar] [CrossRef]
- Brock, J.H.; Arzabe, F.R.; Ortega, F.; Pineiro, A. The effect of limited proteolysis by trypsin and chymotrypsin on bovine colostral IgG1. Immunology 1977, 32, 215–219. [Google Scholar]
- Cleveland, D.W.; Fischer, S.G.; Kirschner, M.W.; Laemmli, U.K. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J. Biol. Chem. 1977, 252, 1102–1106. [Google Scholar] [PubMed]
- Kullmann, W. Kinetics of chymotrypsin- and papain-catalysed synthesis of [leucine]enkephalin and [methionine]enkephalin. Biochem. J. 1984, 220, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, S.; Hovsepyan, M.; Geoghegan, K.F.; Chrunyk, B.A.; Engel, P.C. A thermally sensitive loop in clostridial glutamate dehydrogenase detected by limited proteolysis. J. Biol. Chem. 2003, 278, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; de Laureto, P.P.; Spolaore, B.; Frare, E. Identifying disordered regions in proteins by limited proteolysis. Methods Mol. Biol. 2012, 896, 297–318. [Google Scholar] [PubMed]
- Korotkov, K.V.; Sandkvist, M.; Hol, W.G. The type II secretion system: Biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 2012, 10, 336–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribet, D.; Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015, 17, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Douzi, B.; Ball, G.; Cambillau, C.; Tegoni, M.; Voulhoux, R. Deciphering the Xcp Pseudomonas aeruginosa type II secretion machinery through multiple interactions with substrates. J. Biol. Chem. 2011, 286, 40792–40801. [Google Scholar] [CrossRef] [Green Version]
- Douzi, B.; Durand, E.; Bernard, C.; Alphonse, S.; Cambillau, C.; Filloux, A.; Tegoni, M.; Voulhoux, R. The XcpV/GspI pseudopilin has a central role in the assembly of a quaternary complex within the T2SS pseudopilus. J. Biol. Chem. 2009, 284, 34580–34589. [Google Scholar] [CrossRef] [Green Version]
- Franz, L.P.; Douzi, B.; Durand, E.; Dyer, D.H.; Voulhoux, R.; Forest, K.T. Structure of the minor pseudopilin XcpW from the Pseudomonas aeruginosa type II secretion system. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 124–130. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Faucher, F.; Zhang, W.; Wang, S.; Neville, N.; Poole, K.; Zheng, J.; Jia, Z. Structure-guided disruption of the pseudopilus tip complex inhibits the Type II secretion in Pseudomonas aeruginosa. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carugo, O. How large B-factors can be in protein crystal structures. BMC Bioinform. 2018, 19, 61. [Google Scholar] [CrossRef]
- Malica, C.; Dal Corso, A. Temperature-dependent atomic B factor: An ab initio calculation. Acta Crystallogr. Sect. A Found. Adv. 2019, 75, 624–632. [Google Scholar] [CrossRef]
- Hinsen, K. Structural flexibility in proteins: Impact of the crystal environment. Bioinformatics 2008, 24, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.R.; Porebski, P.J.; Chruszcz, M.; Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin. Drug Discov. 2011, 6, 771–782. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Trincao, J.; Romao, M.J. X-ray crystallography in drug discovery. Methods Mol. Biol. 2009, 572, 31–56. [Google Scholar]
- Zheng, H.; Hou, J.; Zimmerman, M.D.; Wlodawer, A.; Minor, W. The future of crystallography in drug discovery. Expert Opin. Drug Discov. 2014, 9, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Ennifar, E. X-ray crystallography as a tool for mechanism-of-action studies and drug discovery. Curr. Pharm. Biotechnol. 2013, 14, 537–550. [Google Scholar] [CrossRef]
- Nakano, S.; Megro, S.I.; Hase, T.; Suzuki, T.; Isemura, M.; Nakamura, Y.; Ito, S. Computational Molecular Docking and X-ray Crystallographic Studies of Catechins in New Drug Design Strategies. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, L.; Egli, M. In Situ Proteolysis for Crystallization of Membrane Bound Cytochrome P450 17A1 and 17A2 Proteins from Zebrafish. Curr. Protoc. Protein Sci. 2016, 84, 29.16.1–29.16.19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilski, M. Data Processing Programs for Analysis of Diffraction Images of Macromolecular Crystals Recorded using Synchrotron Radiation. Acta Phys. Pol. A 2012, 121, 871–875. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Ginn, H.M.; Stuart, D.I. Recovery of data from perfectly twinned virus crystals revisited. Acta Crystallogr. D Struct. Biol. 2016, 72, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | XcpVWX |
|---|---|
| Space group | P3 |
| Cell dimensions | |
| a, b, c (Å) | 158.1, 158.1, 64.7 |
| α, β, γ (°) | 120, 120, 60 |
| Resolution (Å) | 43.5–2.83 (3.00–2.83) * |
| Rmerge | 13.8 (19.9) |
| CC (1/2) | 99.8 (64.6) |
| I/σ | 10.3 (1.6) |
| Completeness (%) | 99.2 (95.7) |
| Redundancy | 10 |
| Refinement | |
| Resolution (Å) | 43.5–2.83 |
| No. unique reflections | 42465 |
| Rwork/Rfree | 0.21/0.30 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.12 |
| Protein | Orthorhombic 5VTM | Hexagonal 6UTU | ||
|---|---|---|---|---|
| Mol1 | Mol2 | Mol3 | ||
| XcpV | 89–92 | 60–66 71 79 87–90 104–113 | 59–65 87–98 103–112 | 62–65 88–91 106–113 |
| XcpW | 87 | 86 | ||
| XcpX | 67–75 95 263–267 | 67–75 93 | 66–75 92–98 287–291 | 67–75 91–98 161–163 263–266 286–291 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, S.; Jia, Z. In Situ Proteolysis Condition-Induced Crystallization of the XcpVWX Complex in Different Lattices. Int. J. Mol. Sci. 2020, 21, 308. https://doi.org/10.3390/ijms21010308
Zhang Y, Wang S, Jia Z. In Situ Proteolysis Condition-Induced Crystallization of the XcpVWX Complex in Different Lattices. International Journal of Molecular Sciences. 2020; 21(1):308. https://doi.org/10.3390/ijms21010308
Chicago/Turabian StyleZhang, Yichen, Shu Wang, and Zongchao Jia. 2020. "In Situ Proteolysis Condition-Induced Crystallization of the XcpVWX Complex in Different Lattices" International Journal of Molecular Sciences 21, no. 1: 308. https://doi.org/10.3390/ijms21010308
APA StyleZhang, Y., Wang, S., & Jia, Z. (2020). In Situ Proteolysis Condition-Induced Crystallization of the XcpVWX Complex in Different Lattices. International Journal of Molecular Sciences, 21(1), 308. https://doi.org/10.3390/ijms21010308