Biocatalytic Amplification of UV Signal in Capillary Electrophoresis of MicroRNA


Abstract
:1. Introduction
2. Results and Discussion
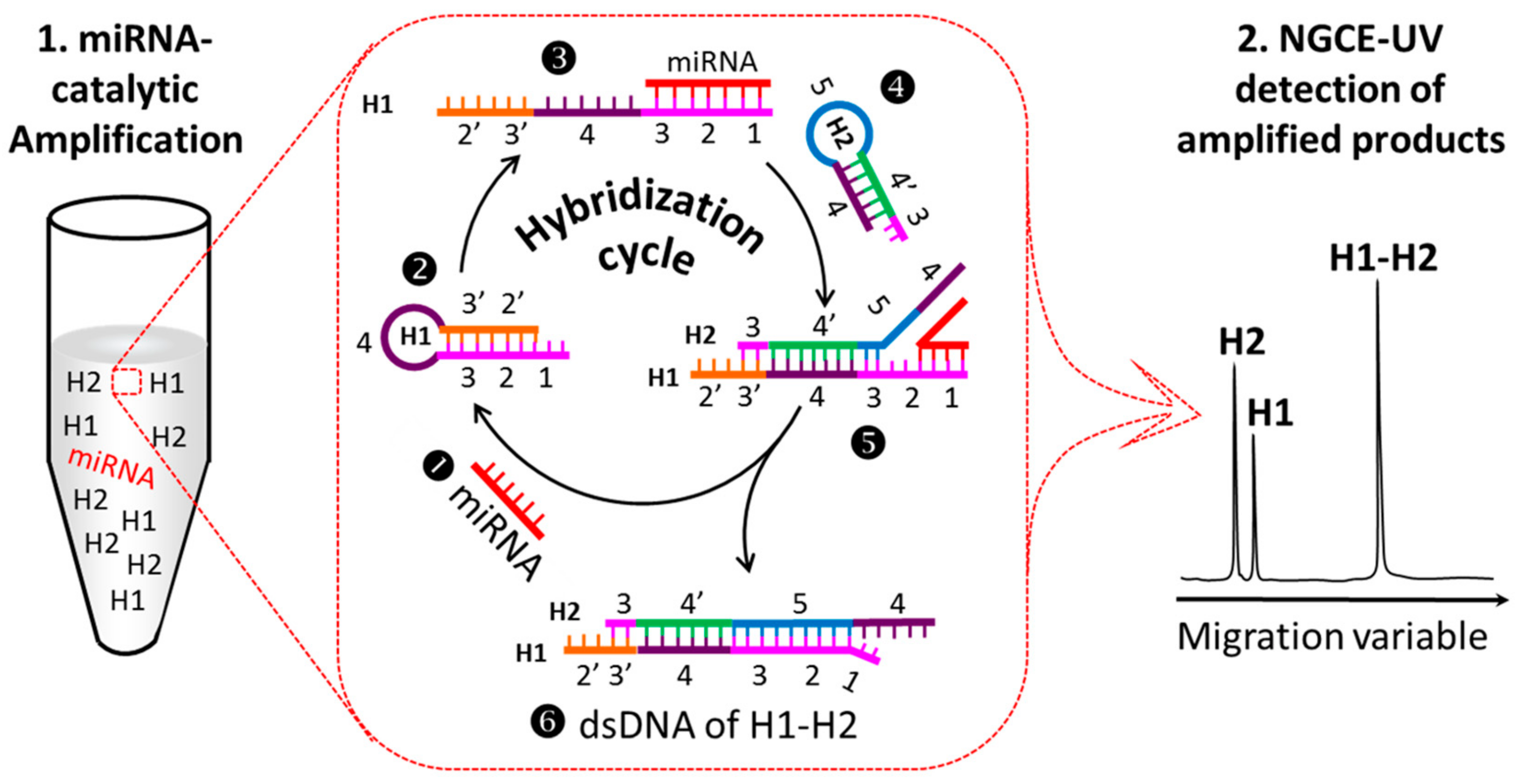
2.1. Principle to Amplify the UV Signals of MiRNAs
2.2. Critical Parameters Associated with the Method
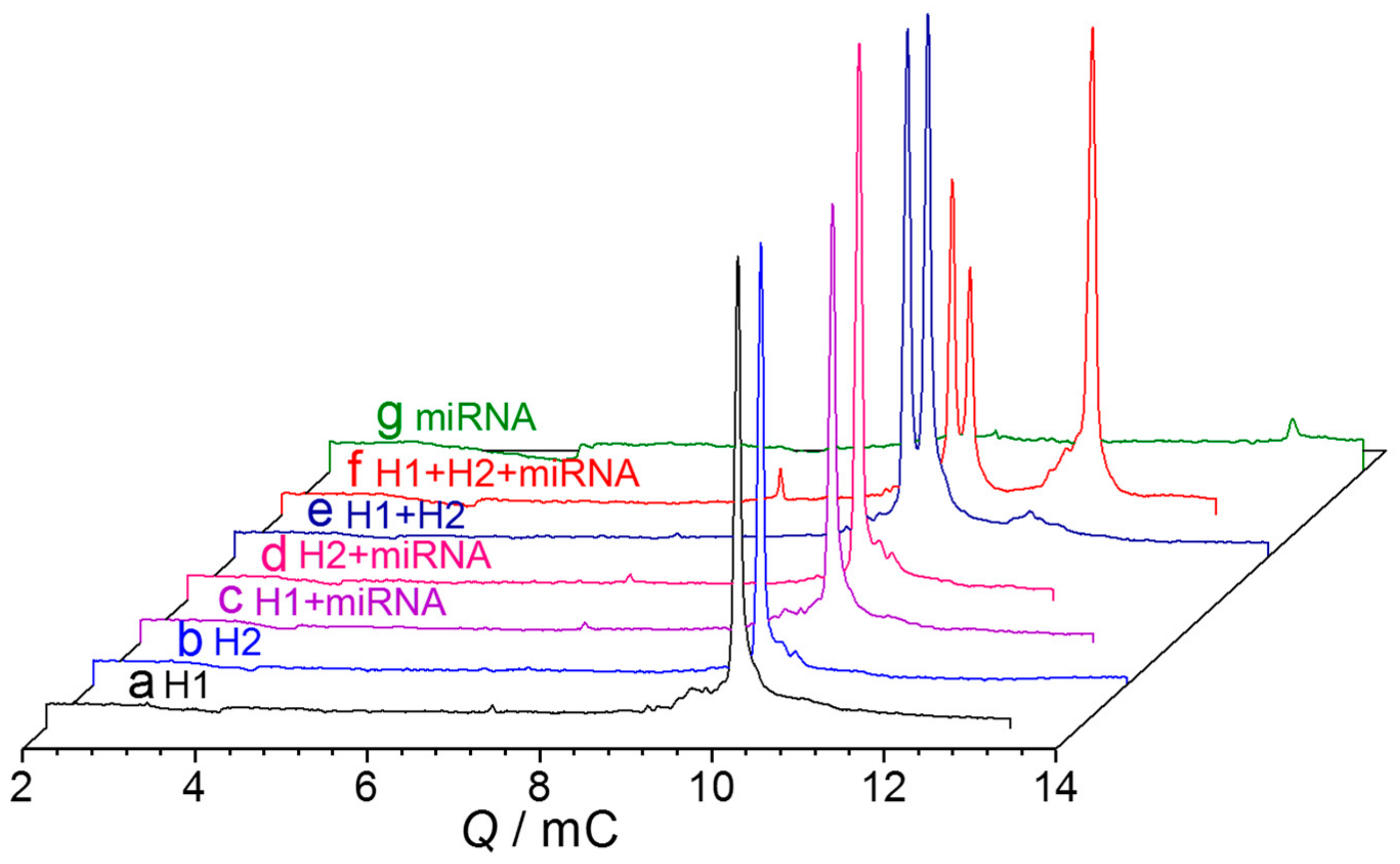
2.2.1. Design and Optimization of H1 and H2 Sequences
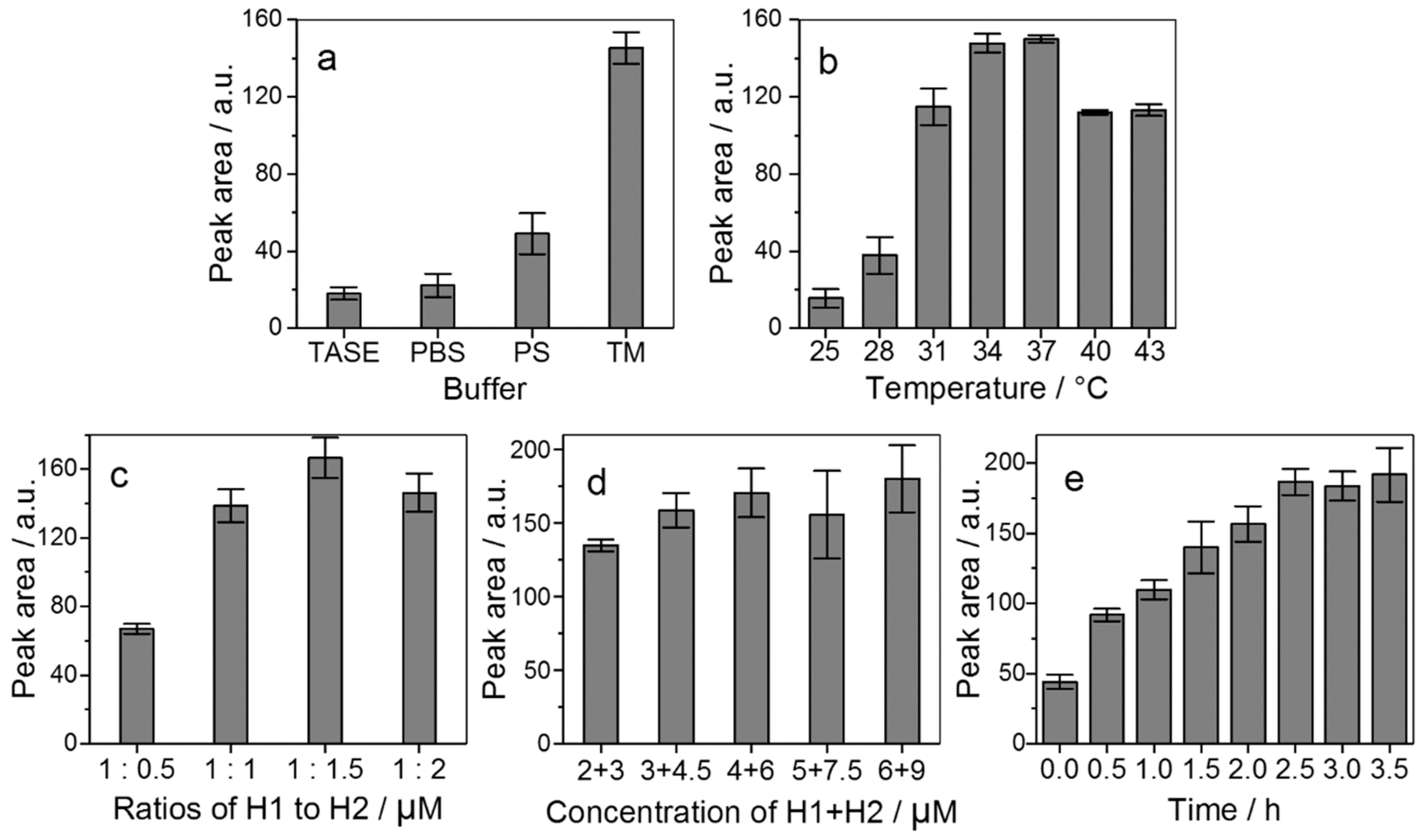
2.2.2. Reaction Conditions
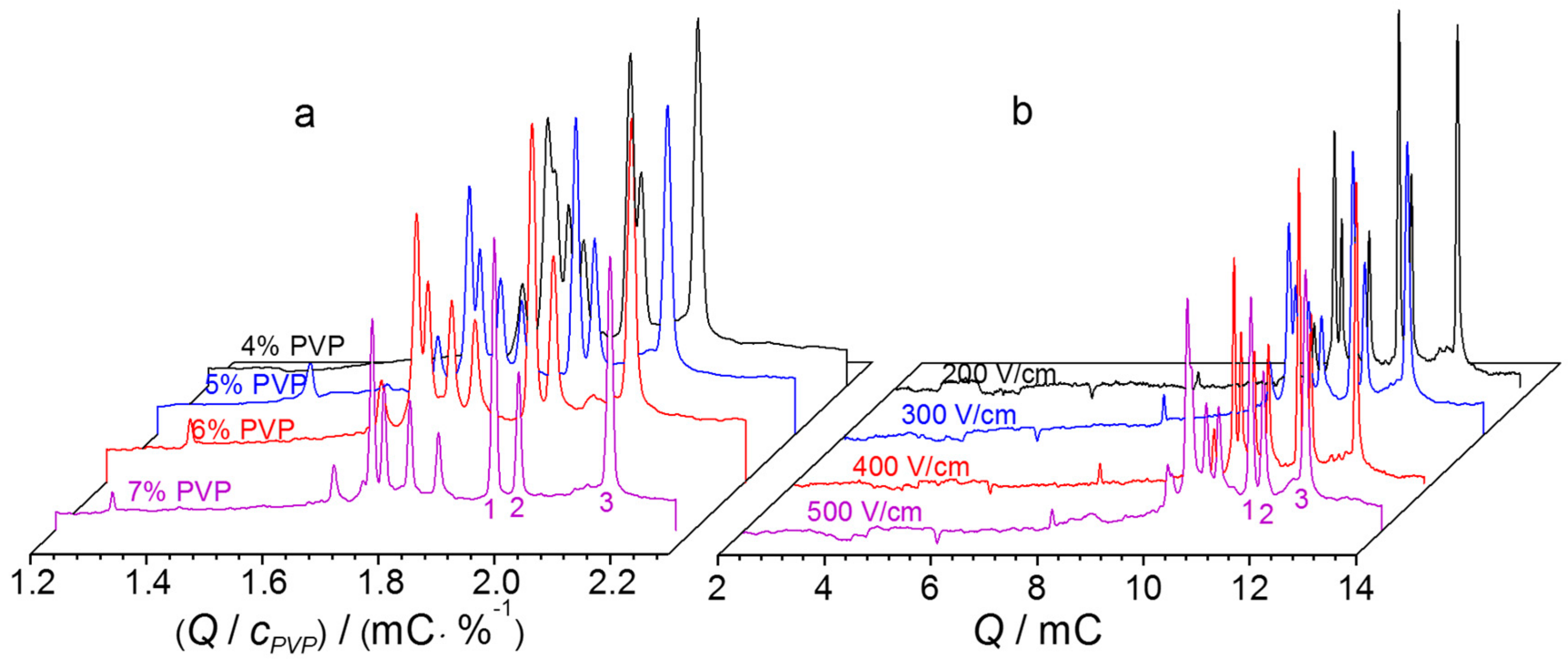
2.2.3. NGCE-UV Conditions
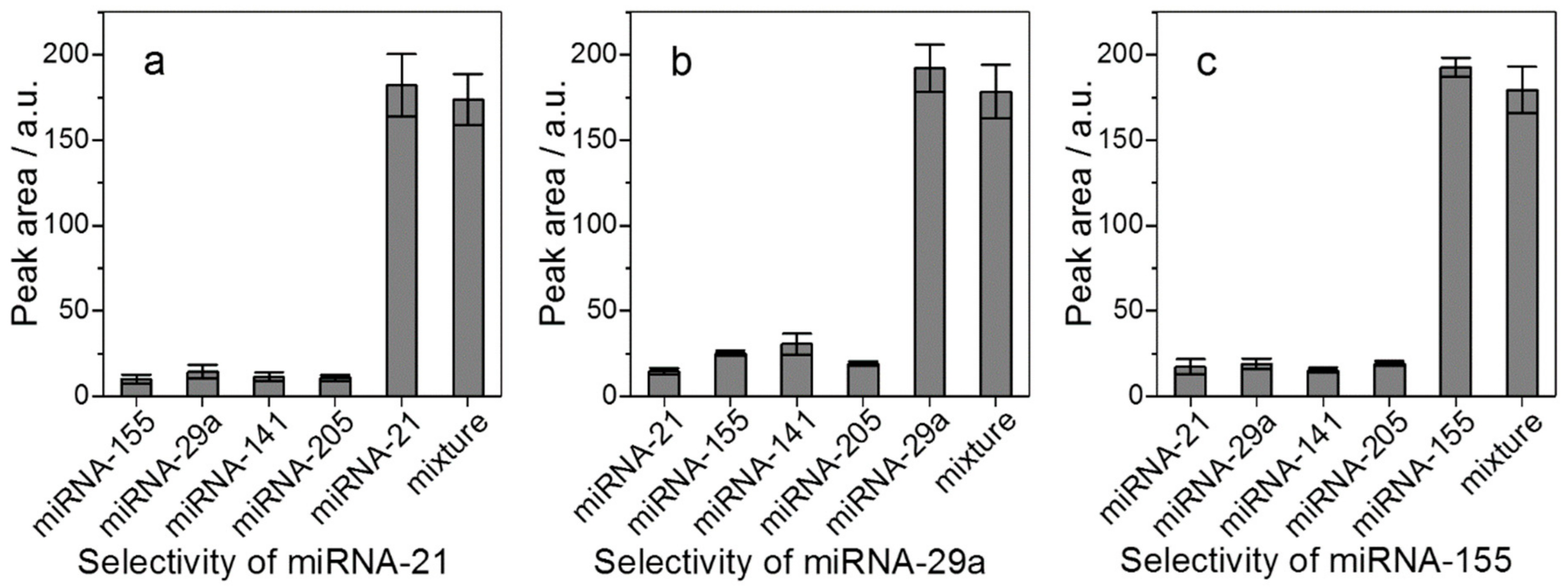
2.3. Method Validation
3. Materials and Methods
3.1. Materials
3.2. Preparation of dsDNAs by MiRNA-Catalyzed Cyclic Hybridization
3.2.1. Preparation of Hairpin DNAs
3.2.2. Hybridization Reaction
3.3. Capillary Electrophoresis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CE | capillary electrophoresis |
| DEPC | diethyl pyrocarbonate |
| LIF | laser-induced fluorescence |
| miRNAs | microRNAs |
| NGCE-UV | non-gel capillary electrophoresis coupled with UV detection |
| PVP | polyvinylpyrrolidone |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| ssDNAs | single-strand DNAs |
| dsDNAs | double-strand DNAs |
References
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef]
- Lin, S.B.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Fan, G.C. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 138–149. [Google Scholar]
- Eacker, S.M.; Dawson, T.M.; Dawson, V.L. Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 2009, 10, 837–841. [Google Scholar] [CrossRef]
- Jin, X.F.; Wu, N.; Wang, L.; Li, J. Circulating MicroRNAs: A novel class of potential biomarkers for diagnosing and prognosing central nervous system diseases. Cell Mol. Neurobiol. 2013, 33, 601–613. [Google Scholar] [CrossRef]
- Wu, Q.; Lu, Z.; Li, H.; Lu, J.; Guo, L.; Ge, Q. Next-generation sequencing of microRNAs for breast cancer detection. J. Biomed. Biotechnol. 2011, 2011, 597145. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Qin, W.; Atasoy, U.; Sauter, E.R. Circulating microRNAs in breast cancer and healthy subjects. BMC Res. Notes. 2009, 2, 89. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.M.; Leung, S.W. Identification of microRNA biomarkers in type 2 diabetes: A meta analysis of controlled profiling studies. Diabetologia 2015, 58, 900–911. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Michael, A.; Bajracharya, S.D.; Yuen, P.S.; Zhou, H.; Star, R.A.; Illei, G.G.; Alevizos, I. Exosomes from human saliva as a source of microRNA biomarkers. Oral. Dis. 2010, 16, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, L.L.; Cao, Y.; Liu, D.; Xu, M.; Liu, H.; Tang, R.N.; Ma, K.L.; Liu, B.C. Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery. Int. J. Biol. Sci. 2013, 9, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; Lin, D.W.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar]
- Zhou, Q.; Li, M.Z.; Wang, X.Y.; Li, Q.Z.; Wang, T.; Zhu, Q.; Zhou, X.C.; Wang, X.; Gao, X.L.; Li, X.W. Immunerelated microRNAs are abundant in breast milk exosomes. Int. J. Biol. Sci. 2012, 8, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Negrini, M.; Ferracin, M.; Sabbioni, S.; Croce, C.M. MicroRNAs in human cancer: From research to therapy. J. Cell Sci. 2007, 120, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Reid, G.; Kirschner, M.B.; Zandwijk, N. Circulating microRNAs: Association with disease and potential use as biomarkers. Crit. Rev. Oncol. Hematol. 2011, 80, 193–208. [Google Scholar] [CrossRef]
- Boyerinas, B.; Park, S.A.; Murmann, A.E.; Peter, M.E. The role of let-7 in cell differentiation and cancer. Endocr. Relat. Cancer 2010, 17, 19–36. [Google Scholar] [CrossRef]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Várallyay, E.; Burgyán, J.; Havelda, Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat. Protoc. 2008, 3, 190–196. [Google Scholar] [CrossRef]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. Bio. Techniques. 2005, 39, 519–525. [Google Scholar] [CrossRef]
- Graybill, R.M.; Bailey, R.C. Emerging biosensing approaches for microRNA analysis. Anal. Chem. 2016, 88, 431–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pall, G.S.; Codony-Servat, C.; Byrne, J.; Ritchie, L.; Hamilton, A. Carbodiimide-mediated Cross-linking of RNA to Nylon Membranes Improves the Detection of siRNA, miRNA and piRNA by Northern blot. Nucleic Acids Res. 2007, 35, e60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroh, E.M.; Parkin, R.K.; Mitchell, P.S.; Tewari, M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods 2010, 50, 298–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.M.; Chang, Y.S.; Chen, S.J.; Chen, J.H.; Chen, H.C.; Chang, P.L. Multiplexed microRNA detection by capillary electrophoresis with laser-induced fluorescence. J. Chromatogr. A 2011, 1218, 2604–2610. [Google Scholar] [CrossRef]
- Ban, E.; Chae, D.K.; Song, E.J. Simultaneous detection of multiple microRNAs for expression profiles of microRNAs in lung cancer cell lines by capillary electrophoresis with dual laser-induced fluorescence. J. Chromatogr. A 2013, 1315, 195–199. [Google Scholar] [CrossRef]
- Wegman, D.W.; Krylov, S.N. Direct quantitative analysis of multiple miRNAs (DQAMmiR). Angew. Chem. Int. Ed. 2011, 50, 10335–10339. [Google Scholar] [CrossRef]
- Wegman, D.W.; Cherney, L.T.; Yousef, G.M.; Krylov, S.N. Universal drag tag for direct quantitative analysis of multiple microRNAs. Anal. Chem. 2013, 85, 6518–6523. [Google Scholar] [CrossRef]
- Li, N.; Jablonowski, C.; Jin, H.; Zhong, W. Stand-alone rolling circle amplification combined with capillary electrophoresis for specific detection of small RNA. Anal. Chem. 2009, 81, 4906–4913. [Google Scholar] [CrossRef]
- Hu, F.C.; Xu, J.Y.; Chen, Y. Surface plasmon resonance imaging detection of sub-femtomolar microRNA. Anal. Chem. 2017, 89, 10071–10077. [Google Scholar] [CrossRef]
- Dirks, R.M.; Pierce, N.A. Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 15275–15278. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.; Choi, H.M.T.; Calvert, C.R.; Pierce, N.A. Programming biomolecular self-assembly pathways. Nature 2008, 451, 318–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Yeung, E.S. A matrix for DNA separation: Genotyping and sequencing using poly (vinylpyrrolidone) solution in uncoated capillaries. Anal. Chem. 1998, 70, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Pang, H.; Yeung, E.S. Simultaneous genetic typing from multiple short tandem repeat loci using a 96-capillary array electrophoresis system. Electrophoresis 1999, 20, 1518–1526. [Google Scholar] [CrossRef]
- Gao, Q.; Yeung, E.S. High-throughput detection of unknown mutations by using multiplexed capillary electrophoresis with poly (vinylpyrrolidone) solution. Anal. Chem. 2000, 72, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.T.; Yeung, E.S. Facilitating data transfer and improving precision in capillary zone electrophoresis with migration indices. Anal. Chem. 1991, 63, 2842–2848. [Google Scholar] [CrossRef]
- Zhao, S.G.; Pan, Z.W.; Chen, X.G.; Hu, Z.D. Analysis of the aconitine alkaloids in traditional Chinese medicines by nonaqueous capillary electrophoresis using a new recording mode. Biomed. Chromatogr. 2004, 18, 381–387. [Google Scholar] [CrossRef]
- Yang, S.H.; Zhang, Y.M.; Liao, T.; Guo, Z.P.; Chen, Y. Electromigration diffusivity spectrometry: A way for simultaneous determination of diffusion coefficients from mixed samples. Electrophoresis 2010, 31, 2949–2956. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Calibration Curve (R2) | Low Concentration (10 nM, n = 3) | Medium Concentration (50 nM, n = 3) | High Concentration (200 nM, n = 3) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intraday RSD (%) | Interday RSD (%) | Intraday Recovery (%) | Intraday RSD (%) | Interday RSD (%) | Intraday Recovery (%) | Intraday RSD (%) | Interday RSD (%) | Intraday Recovery (%) | ||
| miRNA-21 | Y = 2.79c + 24.74 (0.9976) | 2.31 | 9.11 | 88.4 | 3.03 | 2.07 | 99.3 | 4.45 | 1.67 | 95.1 |
| miRNA-29a | Y = 2.72c + 34.05 (0.9948) | 9.66 | 12.61 | 95.0 | 2.50 | 1.21 | 108.6 | 1.39 | 1.97 | 89.9 |
| miRNA-155 | Y = 2.97c + 38.53 (0.9962) | 9.24 | 3.66 | 115.4 | 1.05 | 2.09 | 110.9 | 5.66 | 4.36 | 97.1 |
| Targets | Added Concentration (nM) | Measured Concentration (nM) | RSD (%) (n = 3) | Recovery (%) (n = 3) |
|---|---|---|---|---|
| miRNA-21 | 10 | 10.2 ± 0.5 | 4.79 | 101.8 |
| 50 | 52 ± 2 | 3.55 | 104.1 | |
| 200 | 210 ± 4 | 1.95 | 105.2 | |
| miRNA-29a | 10 | 8.5 ± 0.7 | 8.17 | 85.1 |
| 50 | 56 ± 3 | 4.8 | 112.8 | |
| 200 | 215 ± 12 | 5.37 | 93 | |
| miRNA-155 | 10 | 10.4 ± 0.7 | 6.7 | 104.3 |
| 50 | 53 ± 5 | 9.63 | 105.8 | |
| 200 | 204 ± 4 | 1.87 | 101.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, R.; Chen, Y. Biocatalytic Amplification of UV Signal in Capillary Electrophoresis of MicroRNA. Int. J. Mol. Sci. 2020, 21, 51. https://doi.org/10.3390/ijms21010051
Hu R, Chen Y. Biocatalytic Amplification of UV Signal in Capillary Electrophoresis of MicroRNA. International Journal of Molecular Sciences. 2020; 21(1):51. https://doi.org/10.3390/ijms21010051
Chicago/Turabian StyleHu, Ruibin, and Yi Chen. 2020. "Biocatalytic Amplification of UV Signal in Capillary Electrophoresis of MicroRNA" International Journal of Molecular Sciences 21, no. 1: 51. https://doi.org/10.3390/ijms21010051
APA StyleHu, R., & Chen, Y. (2020). Biocatalytic Amplification of UV Signal in Capillary Electrophoresis of MicroRNA. International Journal of Molecular Sciences, 21(1), 51. https://doi.org/10.3390/ijms21010051