Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis


Abstract
:1. Introduction
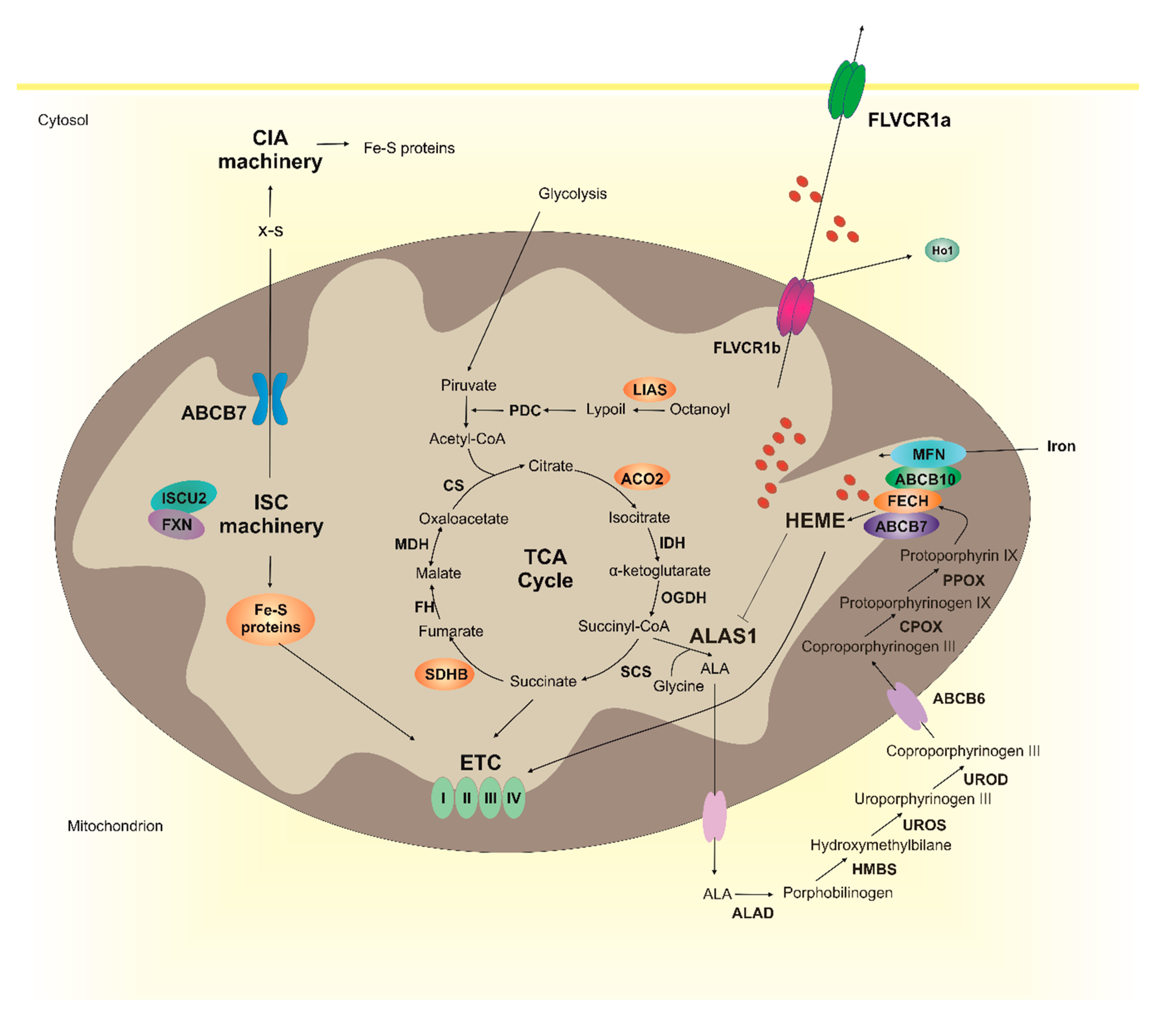
2. Heme Metabolism and Fe-S Cluster Biogenesis
3. Posterior Column Ataxia and Retinitis Pigmentosa (PCARP)
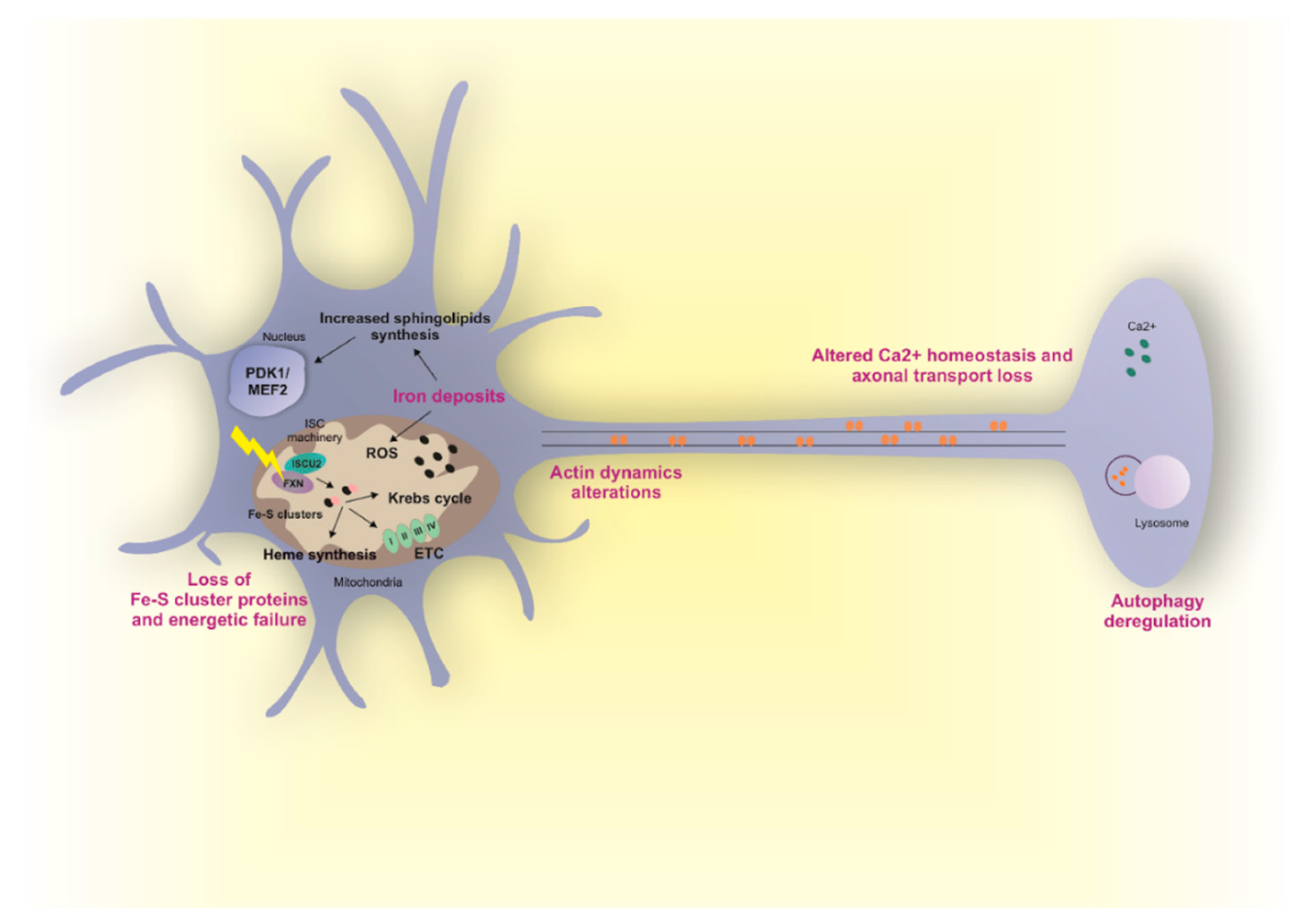
4. Friedreich’s Ataxia (FRDA)
5. X-Linked Sideroblastic Anemia with Ataxia (XLSA/A)
6. Therapeutic Approaches
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akbar, U.; Ashizawa, T. Ataxia. Neurol. Clin. 2015, 33, 225–248. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, J.A.; Luce, J.K. Cerebellar ataxia with weekly 5-fluorouracil administration. Lancet 1971, 1, 138–139. [Google Scholar] [CrossRef]
- Diener, H.C.; Dichgans, J.; Bacher, M.; Guschlbauer, B. Improvement of ataxia in alcoholic cerebellar atrophy through alcohol abstinence. J. Neurol. 1984, 231, 258–262. [Google Scholar] [CrossRef]
- Matthews, B.R.; Jones, L.K.; Saad, D.A.; Aksamit, A.J.; Josephs, K.A. Cerebellar ataxia and central nervous system whipple disease. Arch. Neurol. 2005, 62, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, L.A.; Jarius, S.; Pellkofer, H.L.; Schueller, M.; Krumbholz, M.; Koenig, F.; Johannis, W.; la Fougere, C.; Newman, T.; Vincent, A.; et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J. Neurol. Neurosurg. Psychiatry 2008, 79, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Stephens, K.; Amemiya, A. GeneReviews; Gene Reviews: Seattle, WA, USA, 1993. [Google Scholar]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.J.; Morton, D.H.; Patronas, N.; Nee, L.E. An autosomal recessive disorder with posterior column ataxia and retinitis pigmentosa. Neurology 1997, 49, 1717–1720. [Google Scholar] [CrossRef]
- Higgins, J.J.; Kluetzman, K.; Berciano, J.; Combarros, O.; Loveless, J.M. Posterior column ataxia and retinitis pigmentosa: A distinct clinical and genetic disorder. Mov. Disord. 2000, 15, 575–578. [Google Scholar] [CrossRef]
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. Adv. Exp. Med. Biol. 2019, 1173, 125–143. [Google Scholar] [CrossRef]
- De Michele, G.; Perrone, F.; Filla, A.; Mirante, E.; Giordano, M.; De Placido, S.; Campanella, G. Age of onset, sex, and cardiomyopathy as predictors of disability and survival in Friedreich’s disease: A retrospective study on 119 patients. Neurology 1996, 47, 1260–1264. [Google Scholar] [CrossRef]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef]
- Mollá, B.; Muñoz-Lasso, D.C.; Riveiro, F.; Bolinches-Amorós, A.; Pallardó, F.V.; Fernandez-Vilata, A.; de la Iglesia-Vaya, M.; Palau, F.; Gonzalez-Cabo, P. Reversible Axonal Dystrophy by Calcium Modulation in Frataxin-Deficient Sensory Neurons of YG8R Mice. Front. Mol. Neurosci. 2017, 10, 264. [Google Scholar] [CrossRef]
- Muñoz-Lasso, D.C.; Mollá, B.; Calap-Quintana, P.; García-Giménez, J.L.; Pallardo, F.V.; Palau, F.; Gonzalez-Cabo, P. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci. Rep. 2020, 10, 5207. [Google Scholar] [CrossRef] [Green Version]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Reddi, A.R.; Hamza, I. Heme Mobilization in Animals: A Metallolipid’s Journey. Acc. Chem. Res. 2016, 49, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Harvey, R.M.; Martinez-Guzman, O.; Yuan, X.; Chandrasekharan, B.; Raju, G.; Outten, F.W.; Hamza, I.; Reddi, A.R. Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. USA 2016, 113, 7539–7544. [Google Scholar] [CrossRef] [Green Version]
- Schultz, I.J.; Chen, C.; Paw, B.H.; Hamza, I. Iron and porphyrin trafficking in heme biogenesis. J. Biol. Chem. 2010, 285, 26753–26759. [Google Scholar] [CrossRef] [Green Version]
- Severance, S.; Hamza, I. Trafficking of heme and porphyrins in metazoa. Chem. Rev. 2009, 109, 4596–4616. [Google Scholar] [CrossRef] [Green Version]
- Beinert, H.; Holm, R.H.; Münck, E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020. [Google Scholar] [CrossRef]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stümpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Rouault, T.A. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 2015, 1853, 1493–1512. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Taketani, S.; Kakimoto, K.; Ueta, H.; Masaki, R.; Furukawa, T. Involvement of ABC7 in the biosynthesis of heme in erythroid cells: Interaction of ABC7 with ferrochelatase. Blood 2003, 101, 3274–3280. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematologica 2019, 104, 1756–1767. [Google Scholar] [CrossRef]
- Medlock, A.E.; Shiferaw, M.T.; Marcero, J.R.; Vashisht, A.A.; Wohlschlegel, J.A.; Phillips, J.D.; Dailey, H.A. Identification of the Mitochondrial Heme Metabolism Complex. PLoS ONE 2015, 10, e0135896. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, K.; Sassa, S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J. Clin. Invest. 2000, 105, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.F.; Tchaikovskii, V.; Hoffbrand, A.V.; Fraser, M.E.; Margolis, S. X-linked sideroblastic anemia due to carboxyl-terminal ALAS2 mutations that cause loss of binding to the β-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 2012, 287, 28943–28955. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [Green Version]
- Hanna, D.A.; Martinez-Guzman, O.; Reddi, A.R. Heme Gazing: Illuminating Eukaryotic Heme Trafficking, Dynamics, and Signaling with Fluorescent Heme Sensors. Biochemistry 2017, 56, 1815–1823. [Google Scholar] [CrossRef]
- Galmozzi, A.; Kok, B.P.; Kim, A.S.; Montenegro-Burke, J.R.; Lee, J.Y.; Spreafico, R.; Mosure, S.; Albert, V.; Cintron-Colon, R.; Godio, C.; et al. PGRMC2 is an intracellular haem chaperone critical for adipocyte function. Nature 2019, 576, 138–142. [Google Scholar] [CrossRef]
- Sweeny, E.A.; Singh, A.B.; Chakravarti, R.; Martinez-Guzman, O.; Saini, A.; Haque, M.M.; Garee, G.; Dans, P.D.; Hannibal, L.; Reddi, A.R.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 2018, 293, 14557–14568. [Google Scholar] [CrossRef] [Green Version]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolnek, T.; Zhang, J.; Beardsley, S.; Scheffer, G.L.; Hamza, I. Control of metazoan heme homeostasis by a conserved multidrug resistance protein. Cell Metab. 2014, 19, 1008–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desuzinges-Mandon, E.; Arnaud, O.; Martinez, L.; Huché, F.; Di Pietro, A.; Falson, P. ABCG2 transports and transfers heme to albumin through its large extracellular loop. J. Biol. Chem. 2010, 285, 33123–33133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; Ingoglia, G.; Chiabrando, D.; Mercurio, S.; Turco, E.; Silengo, L.; Altruda, F.; Tolosano, E. Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology 2014, 146, 1325–1338. [Google Scholar] [CrossRef] [Green Version]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe-S cluster biogenesis and its implication in disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. J. Inorg. Biochem. 2018, 183, 107–116. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef]
- Layer, G.; Ollagnier-de Choudens, S.; Sanakis, Y.; Fontecave, M. Iron-sulfur cluster biosynthesis: Characterization of Escherichia coli CYaY as an iron donor for the assembly of [2Fe-2S] clusters in the scaffold IscU. J. Biol. Chem. 2006, 281, 16256–16263. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autréaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Das, D.; Chakrabarti, M.; Lindahl, P.A.; Barondeau, D.P. Frataxin Accelerates [2Fe-2S] Cluster Formation on the Human Fe-S Assembly Complex. Biochemistry 2015, 54, 3880–3889. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef] [Green Version]
- Wingert, R.A.; Galloway, J.L.; Barut, B.; Foott, H.; Fraenkel, P.; Axe, J.L.; Weber, G.J.; Dooley, K.; Davidson, A.J.; Schmid, B.; et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 2005, 436, 1035–1039. [Google Scholar] [CrossRef]
- Pondarré, C.; Antiochos, B.B.; Campagna, D.R.; Clarke, S.L.; Greer, E.L.; Deck, K.M.; McDonald, A.; Han, A.P.; Medlock, A.; Kutok, J.L.; et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet. 2006, 15, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Bekri, S.; Kispal, G.; Lange, H.; Fitzsimons, E.; Tolmie, J.; Lill, R.; Bishop, D.F. Human ABC7 transporter: Gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 2000, 96, 3256–3264. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; Del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Ghezzi, D.; Verrigni, D.; Rizza, T.; Bertini, E.; Martinelli, D.; Zeviani, M.; Singh, A.; Carrozzo, R.; Rouault, T.A. Disease-Causing SDHAF1 Mutations Impair Transfer of Fe-S Clusters to SDHB. Cell Metab. 2016, 23, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Volz, K. The functional duality of iron regulatory protein 1. Curr. Opin. Struct. Biol. 2008, 18, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Eisenstein, R.S. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu. Rev. Nutr. 2000, 20, 627–662. [Google Scholar] [CrossRef]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest. 2010, 120, 1749–1761. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.K.; Dailey, H.A.; Rose, J.P.; Burden, A.; Sellers, V.M.; Wang, B.C. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 2001, 8, 156–160. [Google Scholar] [CrossRef]
- Schneider-Yin, X.; Gouya, L.; Dorsey, M.; Rüfenacht, U.; Deybach, J.C.; Ferreira, G.C. Mutations in the iron-sulfur cluster ligands of the human ferrochelatase lead to erythropoietic protoporphyria. Blood 2000, 96, 1545–1549. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Ghosh, M.C.; Haller, R.G.; Tong, W.H.; Rouault, T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010, 115, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillon, B.; Bulteau, A.L.; Wattenhofer-Donzé, M.; Schmucker, S.; Friguet, B.; Puccio, H.; Drapier, J.C.; Bouton, C. Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. FEBS J. 2009, 276, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Tesfay, L.; Winkler, C.R.; Torti, F.M.; Torti, S.V. Sideroflexin 4 affects Fe-S cluster biogenesis, iron metabolism, mitochondrial respiration and heme biosynthetic enzymes. Sci. Rep. 2019, 9, 19634. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.M.; Greer, J.M.; Ponka, P.; Richardson, D.R. Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: Characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood 2002, 99, 3813–3822. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [Green Version]
- Fiorito, V.; Chiabrando, D.; Tolosano, E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals (Basel) 2018, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Rajadhyaksha, A.M.; Elemento, O.; Puffenberger, E.G.; Schierberl, K.C.; Xiang, J.Z.; Putorti, M.L.; Berciano, J.; Poulin, C.; Brais, B.; Michaelides, M.; et al. Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am. J. Hum. Genet. 2010, 87, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Beigi, F.; Del Pozo-Valero, M.; Martin-Merida, I.; Vahidi Mehrjardi, M.Y.; Manaviat, M.R.; Sherafat, A.; Ayuso, C.; Ghasemi, N. Posterior column ataxia with retinitis pigmentosa (PCARP) in an Iranian patient associated with the. Ophthalmic. Genet. 2020, 41, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Scanga, H.L.; Dansingani, K.K.; Taubenslag, K.J.; Zlotcavitch, L.; Chauhan, B.K.; Sylvester, C.L.; Morton, D.H.; Nischal, K.K. Clinical and imaging characteristics of posterior column ataxia with retinitis pigmentosa with a specific FLVCR1 mutation. Ophthalmic. Genet. 2018, 39, 735–740. [Google Scholar] [CrossRef]
- Shaibani, A.; Wong, L.J.; Wei Zhang, V.; Lewis, R.A.; Shinawi, M. Autosomal recessive posterior column ataxia with retinitis pigmentosa caused by novel mutations in the FLVCR1 gene. Int. J. Neurosci. 2015, 125, 43–49. [Google Scholar] [CrossRef]
- Ishiura, H.; Fukuda, Y.; Mitsui, J.; Nakahara, Y.; Ahsan, B.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; Sakai, T.; Tsuji, S. Posterior column ataxia with retinitis pigmentosa in a Japanese family with a novel mutation in FLVCR1. Neurogenetics 2011, 12, 117–121. [Google Scholar] [CrossRef]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [Green Version]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic. Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef]
- Whelan, L.; Dockery, A.; Wynne, N.; Zhu, J.; Stephenson, K.; Silvestri, G.; Turner, J.; O’Byrne, J.J.; Carrigan, M.; Humphries, P.; et al. Findings from a Genotyping Study of Over 1000 People with Inherited Retinal Disorders in Ireland. Genes (Basel) 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Kuehlewein, L.; Schöls, L.; Llavona, P.; Grimm, A.; Biskup, S.; Zrenner, E.; Kohl, S. Phenotypic spectrum of autosomal recessive retinitis pigmentosa without posterior column ataxia caused by mutations in the FLVCR1 gene. Graefes. Arch. Clin. Exp. Ophthalmol. 2019, 257, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef] [PubMed]
- Bertino, F.; Firestone, K.; Bellacchio, E.; Jackson, K.E.; Asamoah, A.; Hersh, J.; Fiorito, V.; Destefanis, F.; Gonser, R.; Tucker, M.E.; et al. Heme and sensory neuropathy: Insights from novel mutations in the heme exporter FLVCR1. Pain 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castori, M.; Morlino, S.; Ungelenk, M.; Pareyson, D.; Salsano, E.; Grammatico, P.; Tolosano, E.; Kurth, I.; Chiabrando, D. Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, S.; Petrillo, S.; Chiabrando, D.; Bassi, Z.I.; Gays, D.; Camporeale, A.; Vacaru, A.; Miniscalco, B.; Valperga, G.; Silengo, L.; et al. The heme exporter Flvcr1 regulates expansion and differentiation of committed erythroid progenitors by controlling intracellular heme accumulation. Haematologica 2015, 100, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Law, C.J.; Maloney, P.C.; Wang, D.N. Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Scietti, L.; Prajica, A.G.; Bertino, F.; Tolosano, E.; Magnani, F. Expression and purification of the heme exporter FLVCR1a. Protein. Expr. Purif. 2020, 172, 105637. [Google Scholar] [CrossRef]
- Mercurio, S.; Aspesi, A.; Silengo, L.; Altruda, F.; Dianzani, I.; Chiabrando, D. Alteration of heme metabolism in a cellular model of Diamond-Blackfan anemia. Eur. J. Haematol. 2015. [Google Scholar] [CrossRef]
- Rey, M.A.; Duffy, S.P.; Brown, J.K.; Kennedy, J.A.; Dick, J.E.; Dror, Y.; Tailor, C.S. Enhanced alternative splicing of the FLVCR1 gene in Diamond Blackfan anemia disrupts FLVCR1 expression and function that are critical for erythropoiesis. Haematologica 2008, 93, 1617–1626. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef]
- Fiorito, V.; Forni, M.; Silengo, L.; Altruda, F.; Tolosano, E. Crucial role of Flvcr1a in the maintenance of intestinal heme homeostasis. Antioxid. Redox. Signal 2015. [Google Scholar] [CrossRef]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2019, 9, 1540. [Google Scholar] [CrossRef] [Green Version]
- Yanatori, I.; Yasui, Y.; Miura, K.; Kishi, F. Mutations of FLVCR1 in posterior column ataxia and retinitis pigmentosa result in the loss of heme export activity. Blood Cells Mol. Dis. 2012, 49, 60–66. [Google Scholar] [CrossRef]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the Role of Heme in Neurodegeneration. Front. Neurosci. 2018, 12, 712. [Google Scholar] [CrossRef]
- Cossée, M.; Schmitt, M.; Campuzano, V.; Reutenauer, L.; Moutou, C.; Mandel, J.L.; Koenig, M. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: Founder effect and premutations. Proc. Natl. Acad. Sci. USA 1997, 94, 7452–7457. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.E. Friedreich’s ataxia: A clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981, 104, 589–620. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dyck, P.J.; Lambert, E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch. Neurol. 1968, 18, 603–618. [Google Scholar] [CrossRef]
- Said, G.; Marion, M.H.; Selva, J.; Jamet, C. Hypotrophic and dying-back nerve fibers in Friedreich’s ataxia. Neurology 1986, 36, 1292–1299. [Google Scholar] [CrossRef]
- González-Cabo, P.; Palau, F. Mitochondrial pathophysiology in Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 53–64. [Google Scholar] [CrossRef]
- Harding, A.E.; Hewer, R.L. The heart disease of Friedreich’s ataxia: A clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q. J. Med. 1983, 52, 489–502. [Google Scholar]
- Tsou, A.Y.; Paulsen, E.K.; Lagedrost, S.J.; Perlman, S.L.; Mathews, K.D.; Wilmot, G.R.; Ravina, B.; Koeppen, A.H.; Lynch, D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011, 307, 46–49. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef]
- De Michele, G.; Filla, A.; Cavalcanti, F.; Tammaro, A.; Monticelli, A.; Pianese, L.; Di Salle, F.; Perreti, A.; Santoro, L.; Caruso, G.; et al. Atypical Friedreich ataxia phenotype associated with a novel missense mutation in the X25 gene. Neurology 2000, 54, 496–499. [Google Scholar] [CrossRef]
- Cossée, M.; Dürr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschütter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Bartolo, C.; Mendell, J.R.; Prior, T.W. Identification of a missense mutation in a Friedreich’s ataxia patient: Implications for diagnosis and carrier studies. Am. J. Med. Genet. 1998, 79, 396–399. [Google Scholar] [CrossRef]
- Montermini, L.; Richter, A.; Morgan, K.; Justice, C.M.; Julien, D.; Castellotti, B.; Mercier, J.; Poirier, J.; Capozzoli, F.; Bouchard, J.P.; et al. Phenotypic variability in Friedreich ataxia: Role of the associated GAA triplet repeat expansion. Ann. Neurol. 1997, 41, 675–682. [Google Scholar] [CrossRef]
- Castaldo, I.; Pinelli, M.; Monticelli, A.; Acquaviva, F.; Giacchetti, M.; Filla, A.; Sacchetti, S.; Keller, S.; Avvedimento, V.E.; Chiariotti, L.; et al. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 2008, 45, 808–812. [Google Scholar] [CrossRef]
- Evans-Galea, M.V.; Carrodus, N.; Rowley, S.M.; Corben, L.A.; Tai, G.; Saffery, R.; Galati, J.C.; Wong, N.C.; Craig, J.M.; Lynch, D.R.; et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 2012, 71, 487–497. [Google Scholar] [CrossRef]
- Sakamoto, N.; Chastain, P.D.; Parniewski, P.; Ohshima, K.; Pandolfo, M.; Griffith, J.D.; Wells, R.D. Sticky DNA: Self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol. Cell 1999, 3, 465–475. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Bidichandani, S.I. Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 2019, 132, 104606. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Napierala, M.; Puccio, H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Model. Mech. 2012, 5, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 2015, 21, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, J.B.; Côté, M.; Lemieux, B. The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Huang, M.L.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The dentate nucleus in Friedreich’s ataxia: The role of iron-responsive proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.; Li, T.; Li, Z.; Duraine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Ho, T.S.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. Elife 2016, 5. [Google Scholar] [CrossRef]
- Yin, Y.; She, H.; Li, W.; Yang, Q.; Guo, S.; Mao, Z. Modulation of Neuronal Survival Factor MEF2 by Kinases in Parkinson’s Disease. Front. Physiol. 2012, 3, 171. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, J.B. The MEF2 family and the brain: From molecules to memory. Cell Tissue Res. 2013, 352, 179–190. [Google Scholar] [CrossRef]
- Lambert, L.; Bienvenu, T.; Allou, L.; Valduga, M.; Echenne, B.; Diebold, B.; Mignot, C.; Héron, D.; Roth, V.; Saunier, A.; et al. MEF2C mutations are a rare cause of Rett or severe Rett-like encephalopathies. Clin. Genet. 2012, 82, 499–501. [Google Scholar] [CrossRef]
- Poburski, D.; Boerner, J.B.; Koenig, M.; Ristow, M.; Thierbach, R. Time-resolved functional analysis of acute impairment of frataxin expression in an inducible cell model of Friedreich ataxia. Biol. Open 2016, 5, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.; Seznec, H.; Gansmuller, A.; Carelle, N.; Weber, P.; Metzger, D.; Rustin, P.; Koenig, M.; Puccio, H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004, 24, 1987–1995. [Google Scholar] [CrossRef] [Green Version]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Magrane, J.; Rattelle, A.; Stepanova, A.; Galkin, A.; Clark, E.M.; Dong, Y.N.; Halawani, S.M.; Lynch, D.R. Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech. 2017, 10, 1343–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolinches-Amorós, A.; Mollá, B.; Pla-Martín, D.; Palau, F.; González-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodi, R.; Cooper, J.M.; Bradley, J.L.; Manners, D.; Styles, P.; Taylor, D.J.; Schapira, A.H. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc. Natl. Acad. Sci. USA 1999, 96, 11492–11495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutak, R.; Xu, X.; Whitnall, M.; Kashem, M.A.; Vyoral, D.; Richardson, D.R. Proteomic analysis of hearts from frataxin knockout mice: Marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics 2008, 8, 1731–1741. [Google Scholar] [CrossRef]
- Ristow, M.; Pfister, M.F.; Yee, A.J.; Schubert, M.; Michael, L.; Zhang, C.Y.; Ueki, K.; Michael, M.D.; Lowell, B.B.; Kahn, C.R. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12239–12243. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, R.A.; Napoli, E.; Wong, A.; Zhan, S.; Reutenauer, L.; Morin, D.; Buckpitt, A.R.; Taroni, F.; Lonnerdal, B.; Ristow, M.; et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005, 14, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 2009, 18, 2452–2461. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. North. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef]
- Pagon, R.A.; Bird, T.D.; Detter, J.C.; Pierce, I. Hereditary sideroblastic anaemia and ataxia: An X linked recessive disorder. J. Med. Genet. 1985, 22, 267–273. [Google Scholar] [CrossRef]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.; Hellier, K.; Hammans, S.; May, A. X-linked cerebellar ataxia and sideroblastic anaemia associated with a missense mutation in the ABC7 gene predicting V411L. Br. J. Haematol. 2001, 115, 910–917. [Google Scholar] [CrossRef]
- Protasova, M.S.; Grigorenko, A.P.; Tyazhelova, T.V.; Andreeva, T.V.; Reshetov, D.A.; Gusev, F.E.; Laptenko, A.E.; Kuznetsova, I.L.; Goltsov, A.Y.; Klyushnikov, S.A.; et al. Whole-genome sequencing identifies a novel ABCB7 gene mutation for X-linked congenital cerebellar ataxia in a large family of Mongolian ancestry. Eur. J. Hum. Genet. 2016, 24, 550–555. [Google Scholar] [CrossRef] [PubMed]
- D’Hooghe, M.; Selleslag, D.; Mortier, G.; Van Coster, R.; Vermeersch, P.; Billiet, J.; Bekri, S. X-linked sideroblastic anemia and ataxia: A new family with identification of a fourth ABCB7 gene mutation. Eur. J. Paediatr. Neurol. 2012, 16, 730–735. [Google Scholar] [CrossRef]
- Li, J.; Cowan, J.A. Glutathione-coordinated [2Fe-2S] cluster: A viable physiological substrate for mitochondrial ABCB7 transport. Chem. Commun. (CAMB) 2015, 51, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A.; Tong, W.H. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008, 24, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, P.; Biasiotto, G.; Poli, M.; Levi, S.; Verardi, R.; Zanella, I.; Derosas, M.; Ingrassia, R.; Corrado, M.; Arosio, P. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 2007, 109, 3552–3559. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Rouault, T.A. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef]
- Zhang, S.; Napierala, M.; Napierala, J.S. Therapeutic Prospects for Friedreich’s Ataxia. Trends. Pharmacol. Sci. 2019, 40, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Le Quan Sang, K.H.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Nachbauer, W.; Hering, S.; Seifert, M.; Steinkellner, H.; Sturm, B.; Scheiber-Mojdehkar, B.; Reindl, M.; Strasak, A.; Poewe, W.; Weiss, G.; et al. Effects of erythropoietin on frataxin levels and mitochondrial function in Friedreich ataxia--a dose-response trial. Cerebellum 2011, 10, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, C.; Nachbauer, W.; Panzeri, M.; Poewe, W.; Taroni, F.; Boesch, S. Erythropoietin in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 80–87. [Google Scholar] [CrossRef]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014, 20, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Wood, K.M.; Rao, V.P.; Morin, J.; Bhamidipaty, S.; LaBranche, T.P.; Gooch, R.L.; Bozal, F.; Bulawa, C.E.; Guild, B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016, 6, 20019. [Google Scholar] [CrossRef] [Green Version]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Matsui, M.; Corey, D.R. Activating frataxin expression by repeat-targeted nucleic acids. Nat. Commun. 2016, 7, 10606. [Google Scholar] [CrossRef]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef] [PubMed]
- Erwin, G.S.; Grieshop, M.P.; Ali, A.; Qi, J.; Lawlor, M.; Kumar, D.; Ahmad, I.; McNally, A.; Teider, N.; Worringer, K.; et al. Synthetic transcription elongation factors license transcription across repressive chromatin. Science 2017, 358, 1617–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Gene | Function | Neurological Clinical Features | Sites of Pathology | Ref. | |
|---|---|---|---|---|---|
| PCARP | FLVCR1 | Heme export | Sensory ataxia Retinitis pigmentosa | Posterior columns of spinal cord Retina | [8,9] |
| FRDA | FXN | Fe-S cluster biogenesis | Mixed spinocerebellar and sensory ataxiaDysarthriaMuscular weakness Absence of tendon reflexes with deep sensory loss | Large sensory neuronsPosterior columns of spinal cordDentate nucleus of cerebellum | [10,11,12] |
| XLSA/A | ABCB7 | Export of S-X compound from mitochondria | Spinocerebellar ataxia Intention tremor Dysarthria | Cerebellum | [13,14] |
| Mutation(s) | Zygosity | Types of Mutation | Exon/Intron | Ref. |
|---|---|---|---|---|
| c.361A>G (p.Asn121Asp) | Homozygous | Missense | Exon 1 | [79,81] |
| c.721G>A (p.Ala241Thr) | Homozygous | Missense | Exon 1 | [79] |
| c.574T>C (p.Cys192Arg) | Homozygous | Missense | Exon 1 | [79] |
| c.1477G>C (Gly493Arg) | Homozygous | Missense | Exon 8 | [83] |
| c.1547G>A (p.Arg516Gln) c.1593+5 +8delGTAA | Compound Heterozygous | Missense Splicing | Exon 9 Intron 9–10 | [82] |
| c.596T>C (p.Leu199Pro) | Homozygous | Missense | Exon 1 | [80] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiabrando, D.; Bertino, F.; Tolosano, E. Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis. Int. J. Mol. Sci. 2020, 21, 3760. https://doi.org/10.3390/ijms21113760
Chiabrando D, Bertino F, Tolosano E. Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis. International Journal of Molecular Sciences. 2020; 21(11):3760. https://doi.org/10.3390/ijms21113760
Chicago/Turabian StyleChiabrando, Deborah, Francesca Bertino, and Emanuela Tolosano. 2020. "Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis" International Journal of Molecular Sciences 21, no. 11: 3760. https://doi.org/10.3390/ijms21113760
APA StyleChiabrando, D., Bertino, F., & Tolosano, E. (2020). Hereditary Ataxia: A Focus on Heme Metabolism and Fe-S Cluster Biogenesis. International Journal of Molecular Sciences, 21(11), 3760. https://doi.org/10.3390/ijms21113760