An Evidence-Based Update on the Molecular Mechanisms Underlying Periodontal Diseases

 ,
,  and
and 
Abstract
:
{kind=link}
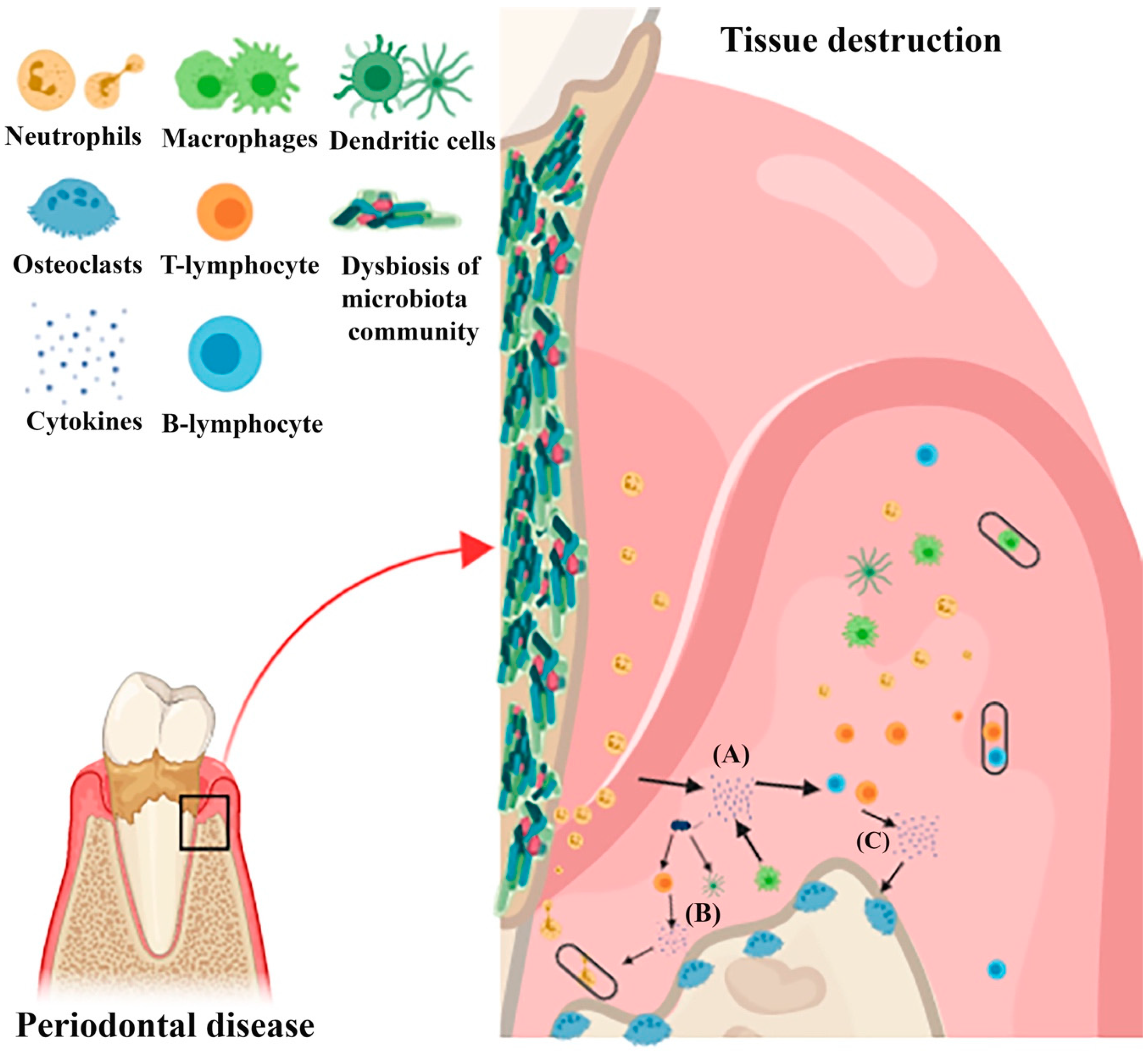{kind=link}
{kind=link}
{kind=link}
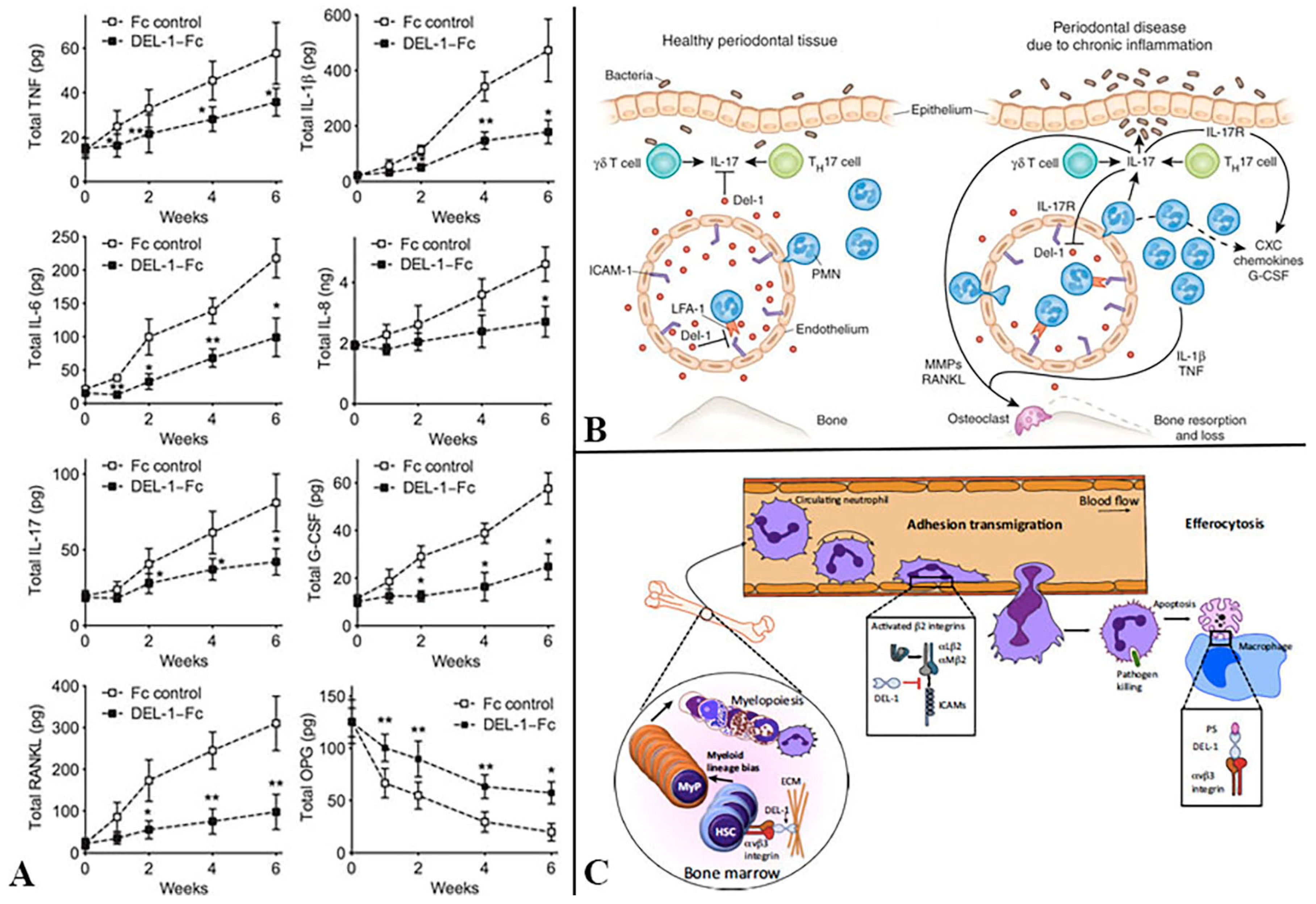{kind=link}
1. Introduction
2. Evolving Concepts in Host Modulation
2.1. Cytokines, Proteases and Prostaglandins
2.2. Pathways of Tissue Destruction via MMPs
2.3. RANK/RANKL/OPG Interactions
2.4. Evidence-Based Updates on RANK, RANKL and OPG
3. Genomic and Transcriptomic Association
Proteomics and Peptidomics Approaches to the Disease
4. Investigations and New Molecules Discovered in Immunochemical Pathways of Periodontitis
5. Clinical Implication
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cells |
| DC | Dendritic cells |
| IL | Interleukin |
| INF | Interferon |
| MMPs | Matrix metalloproteinases |
| OPG | Osteoprotegrin |
| ROS | Reactive oxygen species |
| PDL | Periodontal ligaments |
| PD | Periodontal diseases |
| TNF | Tumor necrosis factor |
| LPS | Lipopolysaccharides |
| TLR | Toll like receptor |
References
- Kassebaum, N.J.; Bernabe, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.L.; Marcenes, W. Global burden of severe periodontitis in 1990-2010: A systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef]
- Richards, D.; Marcenes, W.; Kassebaum, N.J.; Bernabé, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.L. Review finds that severe periodontitis affects 11% of the world population. Evid. Based Dent. 2014, 15, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E.; Bartold, P.M.; Reynolds, E.C. The Nexus Between Periodontal Inflammation and Dysbiosis. Front. Immunol. 2020, 11, 511. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, N.; Arce, R.M. Periodontal inflammation: Integrating genes and dysbiosis. Periodontol. 2000 2020, 82, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Bartold, P.M.; Van Dyke, T.E. Periodontitis: A host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontol. 2000 2013, 62, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Bartold, P.M.; Van Dyke, T.E. Host modulation: controlling the inflammation to control the infection. Periodontol. 2000 2017, 75, 317–329. [Google Scholar] [CrossRef]
- Sulijaya, B.; Takahashi, N.; Yamazaki, K. Host modulation therapy using anti-inflammatory and antioxidant agents in periodontitis: A review to a clinical translation. Arch. of Oral Biol. 2019, 105, 72–80. [Google Scholar] [CrossRef]
- Trindade, F.; Oppenheim, F.G.; Helmerhorst, E.J.; Amado, F.; Gomes, P.S.; Vitorino, R. Uncovering the molecular networks in periodontitis. Proteom. Clin. Appl. 2014, 87, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Kebschull, M.; Demmer, R.T.; Papapanou, P.N. “Gum bug, leave my heart alone!”-epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J. Dent. Res. 2010, 89, 879–902. [Google Scholar] [CrossRef] [Green Version]
- Genco, R.J.; Van Dyke, T.E. Reducing the risk of CVD in patients with periodontitis. Nat. Rev. Cardiol. 2010, 7, 479–480. [Google Scholar] [CrossRef]
- Corbella, S.; Veronesi, P.; Galimberti, V.; Weinstein, R.; Del Fabbro, M.; Francetti, L. Is periodontitis a risk indicator for cancer? A meta-analysis. PLoS ONE 2018, 13, e0195683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingham, C.O.; Moni, M. Periodontal disease and rheumatoid arthritis: The evidence accumulates for complex pathobiologic interactions. Curr. Opin. Rheumatol. 2013, 25, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and diabetes: a two-way relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Sima, C.; Van Dyke, T.E. Systems medicine and periodontal diseases. In Translational Systems Medicine and Oral Disease; Sonis, S.T., Villa, A., Eds.; Elsevier: Amsterdan, NL, USA, 2019; pp. 249–282. [Google Scholar]
- Tettamanti, L.; Gaudio, R.M.; Iapichino, A.; Mucchi, D.; Tagliabue, A. Genetic susceptibility and periodontal disease: A retrospective study on a large italian sample. Oral Implantol. 2017, 10, 20–27. [Google Scholar] [CrossRef]
- Heidari, Z.; Moudi, B.; Mahmoudzadeh-Sagheb, H. Immunomodulatory factors gene polymorphisms in chronic periodontitis: An overview. BMC Oral Health 2019, 19, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, M.K.; De Carvalho, A.C.G.; Alves, E.H.P.; Da Silva, F.R.P.; Pessoa, L.D.S.; Vasconcelos, D.F.P. Genetic Factors and the Risk of Periodontitis Development: Findings from a Systematic Review Composed of 13 Studies of Meta-Analysis with 71,531 Participants. Int. J. Dent. 2017. [Google Scholar] [CrossRef]
- Guzeldemir-Akcakanat, E.; Alkan, B.; Sunnetci-Akkoyunlu, D.; Gurel, B.; Balta, V.M.; Kan, B.; Akgun, E.; Yilmaz, E.B.; Baykal, E.T.; Cine, N.; et al. Molecular signatures of chronic periodontitis in gingiva: A genomic and proteomic analysis. J. Periodontol. 2019, 90, 663–673. [Google Scholar] [CrossRef]
- Kornman, K.S. Mapping the Pathogenesis of Periodontitis: A New Look. J. Periodontol. 2008, 79, 1560–1568. [Google Scholar] [CrossRef]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol. 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Buduneli, N. Biomarkers in Periodontal Health and Disease; Springer International Publishing: Basel, Switzerland, 2020; Nature of Periodontal Diseases; pp. 9–19. [Google Scholar]
- Bengtsson, Å.K.; Ryan, E.J. Immune function of the decoy receptor osteoprotegerin. Crit. Rev. Immunol. 2002, 22, 201–215. [Google Scholar] [PubMed]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro Souto, G.; Queiroz, C.M.; De Abreu, M.H.N.G.; Costa, F.O.; Mesquita, R.A. Pro-inflammatory, Th1, Th2, Th17 cytokines and dendritic cells: A cross-sectional study in chronic periodontitis. PLoS ONE 2014, 9. [Google Scholar]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Guglani, L.; Khader, S.A. Th17 cytokines in mucosal immunity and inflammation. Curr. Opin. HIV AIDS 2010, 5, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign bodies or tuners for the immune response? Int. J. Mol. Sci. 2019, 20, 636. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Braswell, L.D.; Collins, J.G.; Boyd, D.L.; Jeffcoat, M.K.; Reddy, M.; Li, K.L.; Wilensky, S.; Vogel, R.; Alfano, M. Changes in inflammatory mediators in experimental periodontitis in the rhesus monkey. Infect. Immun. 1993, 61, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Albrektsson, T.; Jemt, T.; Mölne, J.; Tengvall, P.; Wennerberg, A. On inflammation-immunological balance theory—A critical apprehension of disease concepts around implants: Mucositis and marginal bone loss may represent normal conditions and not necessarily a state of disease. Clin. Implant. Dent. Relat. Res. 2019, 21, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. New developments in neutrophil biology and periodontitis. Periodontol. 2000 2020, 82, 78–92. [Google Scholar] [CrossRef]
- Morinha, F.; Albuquerque, C.; Requicha, J.; Dias, I.; Leitao, J.; Gut, I.; Guedes-Pinto, H.; Viegas, C.; Baston, E. Detection and characterization of interleukin-6 gene variants in Canis familiaris: association studies with periodontal disease. Gene 2011, 485, 139–145. [Google Scholar] [CrossRef]
- Offenbacher, S.; Barros, S.P.; Paquette, D.W.; Winston, J.L.; Biesbrock, A.R.; Thomason, R.G.; Gibb, R.G.; Fulmer, A.W.; Tiesman, J.P.; Juhlin, K.D.; et al. Gingival Transcriptome Patterns During Induction and Resolution of Experimental Gingivitis in Humans. J. Periodontol. 2009, 80, 1963–1982. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. Osteoclast differentiation and activation. Clin. Calcium 2007, 17, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Menale, C.; Villa, A. The RANKL-RANK axis: A bone to thymus round trip. Front. Immunol. 2019, 10, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kohli, M.; Zhou, Q.; Graves, D.T.; Amar, S. Short- and Long-Term Effects of IL-1 and TNF Antagonists on Periodontal Wound Healing. J. Immunol. 2004, 173, 3514–3523. [Google Scholar] [CrossRef]
- Rath-Deschner, B.; Memmert, S.; Damanaki, A.; Nokhbehsaim, M.; Eick, S.; Cirelli, J.A.; Götz, W.; Deschner, J.; Jäger, A.; Nogueira, A.V.B. CXCL1, CCL2, and CCL5 modulation by microbial and biomechanical signals in periodontal cells and tissues- in vitro and in vivo studies. Clin. Oral Investig. 2020, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Girnary, M.; Wang, L.; Jiao, Y.; Zeng, E.; Mercer, K.; Zhang, J.; Marchesan, J.T.; Yu, N.; Moss, K.; et al. IL-10 Dampens an IL-17–Mediated Periodontitis-Associated Inflammatory Network. J. Immunol. 2020, ji1900532. [Google Scholar] [CrossRef]
- Xue, M.; Jackson, C.J. Extracellular Matrix Reorganization During Wound Healing and Its Impact on Abnormal Scarring. Adv. Wound Care. 2015, 4, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Rowley, A.T.; Nagalla, R.R.; Wang, S.W.; Liu, W.F. Extracellular Matrix-Based Strategies for Immunomodulatory Biomaterials Engineering. Adv. Healthc. Mater. 2019, 8, 1801578. [Google Scholar] [CrossRef]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate Functions of Matrix Metalloproteinases in Physiological and Pathological Conditions. J. Cell. Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44–46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Birkedal-Hansen, H.; Moore, W.G.I.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix metalloproteinases: A review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanti, L.; Heino, J.; López-Otín, C.; Kähäri, V.M. Induction of collagenase-3 (MMP-13) expression in human skin fibroblasts by three-dimensional collagen is mediated by p38 mitogen-activated protein kinase. J. Biol. Chem. 1999, 274, 2446–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, L.M.; Lee, H.M.; Greenwald, R.A.; Ryan, M.E.; Sorsa, T.; Salo, T.; Gianobile, W.V. A matrix metalloproteinase inhibitor reduces bone-type collagen degradation fragments and specific collagenases in gingival crevicular fluid during adult periodontitis. Inflamm. Res. 1997, 46, 310–319. [Google Scholar] [CrossRef]
- Infante, M.; Fabi, A.; Cognetti, F.; Gorini, S.; Caprio, M.; Fabbri, A. RANKL/RANK/OPG system beyond bone remodeling: Involvement in breast cancer and clinical perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kearns, A.E.; Khosla, S.; Kostenuik, P.J. Receptor activator of nuclear factor κB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocrine Rev. 2008, 29, 155–192. [Google Scholar] [CrossRef]
- Nakashima, T.; Kobayashi, Y.; Yamasaki, S.; Kawakami, A.; Eguchi, K.; Sasaki, H.; Sakai, H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-κB ligand: Modulation of the expression by osteotropic factors and cytokines. Biochem. Biophys. Res. Commun. 2000, 275, 768–775. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [Green Version]
- Sojod, B.; Chateau, D.; Mueller, C.G.; Babajko, S.; Berdal, A.; Lézot, F.; Castaneda, B. RANK/RANKL/OPG signalization implication in periodontitis: New evidence from a RANK transgenic mouse model. Front. Physiol. 2017, 8, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paknejad, M.; Sattari, M.; Roozbahani, Z.; Ershadi, M.; Mehrfard, A. Relationships between high-mobility group protein b1 and triggering receptor expressed on myeloid cells concentrations in gingival crevicular fluid and chronic periodontitis. Iran. J. Allergy Asthma Immunol. 2016, 15, 381–385. Available online: http://ijaai.tums.ac.ir/index.php/ijaai/article/view/664 (accessed on 7 April 2020). [PubMed]
- Yuce, H.B.; Gokturk, O.; Turkal, H.A.; Inanir, A.; Benli, I.; Demir, O. Assessment of local and systemic 25-hydroxy-vitamin D, RANKL, OPG, and TNF levels in patients with rheumatoid arthritis and periodontitis. J. Oral Sci. 2017, 59, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C. Molecular pathways: Osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin. Cancer Res. 2012, 18, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Atala, A. Re: Testosterone replacement effectively inhibits the development of experimental autoimmune orchitis in rats: Evidence for a direct role of testosterone on regulatory T cell expansion. J. Urol. 2012, 187, 351. [Google Scholar] [CrossRef]
- Takeichi, O.; Haber, J.; Kawai, T.; Smith, D.J.; Moro, I.; Taubman, M.A. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J. Dent. Res. 2000, 79, 1548–1555. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.; Sarmadi, M.; Eskan, M.A.; Wild, T.; Dutzan, N.; Abusleme, L.; Zenobia, C.; Hosue, K.; Abe, T.; et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 2014, 6, 229ra40. [Google Scholar] [CrossRef] [Green Version]
- Qasim, S.S.B.; Zafar, M.S.; Niazi, F.H.; Alshahwan, M.; Omar, H.; Daood, U. Functionally Graded Biomimetic Biomaterials in Dentistry: An Evidence-Based Update. J. Biomater. Sci. Polym. Ed. 2020, 1–20. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Diaz, P.I.; Hoare, A.; Hong, B.-Y. Subgingival Microbiome Shifts and Community Dynamics in Periodontal Diseases. J. Calif. Dent. Assoc. 2016, 44, 421–435. [Google Scholar]
- Kim, Y.G.; Kim, M.; Kang, J.H.; Kim, H.J.; Park, J.W.; Lee, J.M.; Abusleme, L.; Zenobia, C.; Hosur, K.B.; Abe, T.; et al. Transcriptome sequencing of gingival biopsies from chronic periodontitis patients reveals novel gene expression and splicing patterns. Hum. Genom. 2016, 10, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, C.; Moll, R.; Kubicka, S.; Ouhayoun, J.P.; Franke, W.W. Characterization of human cytokeratin 2, an Epidermal cytoskeletal protein synthesized late during differentiation. Exp. Cell. Res. 1992, 202, 132–141. [Google Scholar] [CrossRef]
- Degrassi, G.; Aguilar, C.; Bosco, M.; Zahariev, S.; Pongor, S.; Venturi, V. Plant growth-promoting Pseudomonas putida WCS358 produces and secretes four cyclic dipeptides: Cross-talk with quorum sensing bacterial sensors. Curr. Microbiol. 2002, 45, 250–254. [Google Scholar] [CrossRef]
- Abusleme, L.; Dupuy, A.K.; Dutzan, N.; Silva, N.; Burleson, J.A.; Strausbaugh, L.D.; Gamonal, J.; Diaz, P.I. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Divaris, K.; Moss, K.; Yu, N.; Barros, S.; Marchesan, J.; Morelli, T.; Agler, C.; Kim, S.J.; Wu, D.; et al. The novel ASIC2 locus is associated with severe gingival inflammation. JDR Clin. Transl. Res. 2016, 1, 163–170. [Google Scholar] [CrossRef]
- Holden, M.T.G.; Chhabra, S.R.; De Nys, R.; Stead, P.; Bainton, N.J.; Hill, P.J.; Manefield, M.; Kumar, N.; Labatte, M.; England, D.; et al. Quorum-sensing cross talk: Isolation and chemical characterization of cyclic dipeptides from Pseudomonas aeruginosa and other Gram-negative bacteria. Mol. Microbiol. 1999, 33, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the human oral microbiome during health and disease. mBio 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Misra, S. Human gene therapy: a brief overview of the genetic revolution. J. Assoc. Physicians India 2013, 61, 127–133. [Google Scholar]
- Siddique, N.; Raza, H.; Ahmed, S.; Khurshid, Z.; Zafar, M.S. Gene therapy: A paradigm shift in dentistry. Genes 2016, 7, 98. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, P.K.T.; Jeng, L.B.; Huang, C.S.; Yang, L.C.; Chung, H.Y.; Chang, S.C.-N. Periodontal regeneration using ex vivo autologous stem cells engineered to express the BMP-2 gene: An alternative to alveolaplasty. Gene Ther. 2008, 15, 1469–1477. [Google Scholar] [CrossRef]
- Schaefer, A.S.; Richter, G.M.; Nothnagel, M.; Manke, T.; Dommisch, H.; Jacobs, G.; Arlt, A.; Rosenstiel, P.; Noack, B.; Groesnner-Schreiber, B.; et al. A genome-wide association study identifies GLT6D1 as a susceptibility locus for periodontitis. Hum. Mol. Genet. 2009, 9, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Willenborg, C.; Richter, G.M.; Jockel-Schneider, Y.; Graetz, C.; Staufenbiel, I.; Wellman, J.; Berger, K.; Krone, B.; Hoffman, P.; et al. A genome-wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Hum. Mol. Genet. 2017, 26, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Okada, Y.; Ozaki, K.; Urayama, K.; Kanai, M.; Kobayashi, H.; Gokyu, M.; Izumi, Y.; Tanaka, T. Association of NOD2 Mutations with Aggressive Periodontitis. J. Dent. Res. 2017, 96, 1100–1105. [Google Scholar] [CrossRef]
- Hong, K.W.; Shin, M.S.; Ahn, Y.B.; Lee, H.J.; Kim, H.D. Genomewide association study on chronic periodontitis in Korean population: Results from the Yangpyeong health cohort. J. Clin. Periodontol. 2015, 42, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.E.; Sofer, T.; Wong, Q.; Kerr, K.F.; Agler, C.; Shaffer, J.R.; Beck, J.D.; Offenbacher, S.; Salazar, C.R.; North, K.E.; et al. Chronic Periodontitis Genome-wide Association Study in the Hispanic Community Health Study / Study of Latinos. J. Dent. Res. 2017, 96, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teumer, A.; Holtfreter, B.; Völker, U.; Petersmann, A.; Nauck, M.; Biffar, R.; Völzke, H.; Kroemer, H.K.; Meisal, P.; Homuth, G.; et al. Genome-wide association study of chronic periodontitis in a general German population. J. Clin. Periodontol. 2013, 40, 977–985. [Google Scholar] [CrossRef]
- Hiyari, S.; Green, E.; Pan, C.; Lari, S.; Davar, M.; Davis, R.; Camargo, P.M.; Tetradis, S.; Lusis, A.J.; Pirih, F.Q. Genomewide Association Study Identifies Cxcl Family Members as Partial Mediators of LPS-Induced Periodontitis. J. Bone Miner. Res. 2018, 33, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Demmer, R.T.; Behle, J.H.; Wolf, D.L.; Handfield, M.; Kebschull, M.; Celenti, R.; Pavlidis, P.; Papapanou, P.N. Transcriptomes in Healthy and Diseased Gingival Tissues. J. Periodontol. 2008, 79, 2112–2124. [Google Scholar] [CrossRef]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Louten, J.; Boniface, K.; de Waal Malefyt, R. Development and function of TH17 cells in health and disease. J. Allergy Clin. Immunol. 2009, 123, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Human Th17 cells in infection and autoimmunity. Microbes Infect. 2009, 11, 620–624. [Google Scholar] [CrossRef]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, M.; Ghosh, S.; Vishveshwara, S. Protein Structure and Function: Looking through the Network of Side-Chain Interactions. Curr. Protein Pept. Sci. 2015, 17, 4–25. [Google Scholar] [CrossRef]
- Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 1998, 92, 291–294. [Google Scholar] [CrossRef] [Green Version]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Nguyen, T.; Sedghi, L.; Ganther, S.; Malone, E.; Kamarajan, P.; Kapila, Y.L. Host-microbe interactions: Profiles in the transcriptome, the proteome, and the metabolome. Periodontol. 2000 2020, 82, 115–128. [Google Scholar] [CrossRef]
- Wittmann-Liebold, B.; Graack, H.R.; Pohl, T. Two-dimensional gel electrophoresis as tool for proteomics studies in combination with protein identification by mass spectrometry. Proteomics 2006, 6, 4688–4703. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xie, Y.; Ramachandran, P.; Loo, R.R.O.; Li, Y.; Loo, J.A.; Wong, D.T. Large-scale identification of proteins in human salivary proteome by liquid chromatography/mass spectrometry and two-dimensional gel electrophoresis-mass spectrometry. Proteomics 2005, 5, 1714–1728. [Google Scholar] [CrossRef]
- Rigaut, G.; Shevchenko, A.; Rutz, B.; Wilm, M.; Mann, M.; Seraphin, B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999, 17, 1030–1032. [Google Scholar] [CrossRef]
- Cuatrecasas, P. Protein purification by affinity chromatography. Derivatizations of agarose and polyacrylamide beads. J. Biol. Chem. 1970, 245, 3059–3065. [Google Scholar] [PubMed]
- Zafar, M.S.; Belton, D.J.; Hanby, B.; Kaplan, D.L.; Perry, C.C. Functional material features of Bombyx mori silk light versus heavy chain proteins. Biomacromolecules 2015, 16, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Khurshid, Z.; Moin, S.F.; Khan, R.S.; Agwan, M.A.S.; Alwadaani, A.H.; Zafar, M.S. Human salivary protein extraction from RNAPro·SALTM, Pure·SALTM, and passive drooling method. Eur. J. Dent. 2017, 11, 385–389. [Google Scholar]
- Berndt, P.; Hobohm, U.; Langen, H. Reliable automatic protein identification from matrix-assisted laser desorption/ionization mass spectrometric peptide fingerprints. Electrophoresis 1999, 3521–3526. [Google Scholar] [CrossRef]
- Kussmann, M.; Nordhoff, E.; Rahbek-Nielsen, H.; Haebel, S.; Rossel-Larsen, M.; Jakobsen, L.; Gobom, J.; Mirgorodskaya, E.; Kroll-Kristensen, A.; Palmll, L.; et al. Matrix-assisted laser desorption/ionization mass spectrometry sample preparation techniques designed for various peptide and protein analytes. J. Mass. Spectrom. 1997, 32, 593–601. [Google Scholar] [CrossRef]
- Zia, K.; Siddiqui, T.; Ali, S.; Farooq, I.; Zafar, M.S.; Khurshid, Z. Nuclear Magnetic Resonance Spectroscopy for Medical and Dental Applications: A Comprehensive Review. Eur. J. Dent. 2019, 13, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, A.; Grzesiek, S. Methodological Advances in Protein NMR. Acc. Chem. Res. 1993, 26, 131–138. [Google Scholar] [CrossRef]
- Bushey, M.M.; Jorgenson, J.W. Automated Instrumentation for Comprehensive Two-Dimensional High-Performance Liquid Chromatography of Proteins. Anal. Chem. 1990, 62, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Horváth, C. High-speed high-performance liquid chromatography of peptides and proteins. J. Chromatogr. A 1995, 705, 3–20. [Google Scholar] [CrossRef]
- Teles, R.; Teles, F.; Frias-Lopez, J.; Paster, B.; Haffajee, A. Lessons learned and unlearned in periodontal microbiology. Periodontol. 2000 2013, 62, 95–162. [Google Scholar] [CrossRef]
- Gonçalves, L.D.R.; Soares, M.R.; Nogueira, F.C.S.; Garcia, C.; Camisasca, D.R.; Domont, G.; Feitosa, A.C.R.; Perieira, D.; Zingali, R.B.; Alves, G. Comparative proteomic analysis of whole saliva from chronic periodontitis patients. J. Proteom. 2010, 73, 1334–1341. [Google Scholar]
- Mizuno, N.; Niitani, M.; Shiba, H.; Iwata, T.; Hayashi, I.; Kawaguchi, H.; Kurihara, H. Proteome analysis of proteins related to aggressive periodontitis combined with neutrophil chemotaxis dysfunction. J. Clin. Periodontol. 2011, 38, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Haigh, B.J.; Stewart, K.W.; Whelan, J.R.K.; Barnett, M.P.G.; Smolenski, G.A.; Wheeler, T.T. Alterations in the salivary proteome associated with periodontitis. J. Clin. Periodontol. 2010, 37, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Teles, R.P.; Gursky, L.C.; Faveri, M.; Rosa, E.A.; Teles, F.R.F.; Feres, M.; Socransky, S.S.; Haffajee, A.D. Relationships between subgingival microbiota and GCF biomarkers in generalized aggressive periodontitis. J. Clin. Periodontol. 2010, 37, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, S.P.; Williams, R.; Offenbacher, S.; Morelli, T. Gingival crevicular fluid as a source of biomarkers for periodontitis. Periodontol. 2000 2016, 70, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, C.; Bellei, E.; Pellacani, C.; Ferrari, D.; Lucchi, A.; Cuoghi, A.; Bergamini, S.; Cortellini, P.; Tomasi, A.; Zaffe, D.; et al. Non-bacterial protein expression in periodontal pockets by proteome analysis. J. Clin. Periodontol. 2013, 40, 573–582. [Google Scholar] [CrossRef]
- Monari, E.; Cuoghi, A.; Bellei, E.; Bergamini, S.; Lucchi, A.; Tomasi, A.; Cortellini, P.; Zaffe, D.; Bertoldi, C. Analysis of protein expression in periodontal pocket tissue: A preliminary study. Proteome Sci. 2015, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Chapple, I.L.C. Periodontal diagnosis and treatment - Where does the future lie? Periodontol. 2000 2009, 51, 9–24. [Google Scholar] [CrossRef]
- Al-Tarawneh, S.K.; Border, M.B.; Dibble, C.F.; Bencharit, S. Defining salivary biomarkers using mass spectrometry-based proteomics: A systematic review. OMICS 2011, 15, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Guzman, Y.A.; Sakellari, D.; Arsenakis, M.; Floudas, C.A. Proteomics for the discovery of biomarkers and diagnosis of periodontitis: A critical review. Expert Rev. Proteom. 2014, 11, 31–41. [Google Scholar] [CrossRef]
- Bostanci, N.; Heywood, W.; Mills, K.; Parkar, M.; Nibali, L.; Donos, N. Application of label-free absolute quantitative proteomics in human gingival crevicular fluid by LC/MSE (Gingival Exudatome). J. Proteome Res. 2010, 9, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, L.H.; Veith, P.D.; Chen, Y.Y.; Chen, D.; Darby, I.B.; Reynolds, E.C. Mass spectrometric analyses of peptides and proteins in human gingival crevicular fluid. J. Proteome Res. 2010, 9, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Huynh, A.H.S.; Veith, P.D.; Mcgregor, N.R.; Adams, G.G.; Chen, D.; Reynolds, E.C.; Ngo, L.H.; Darby, I.B. Gingival crevicular fluid proteomes in health, gingivitis and chronic periodontitis. J. Periodontal Res. 2015, 50, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Hartenbach, F.A.R.R.; Velasquez, É.; Nogueira, F.C.S.; Domont, G.B.; Ferreira, E.; Colombo, A.P.V. Proteomic analysis of whole saliva in chronic periodontitis. J. Proteom. 2020, 213, 103602. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.S.; Kim, Y.G.; Shin, Y.J.; Ko, B.J.; Kim, S.; Kim, H.D. Deep sequencing salivary proteins for periodontitis using proteomics. Clin. Oral Investig. 2019, 23, 3571–3580. [Google Scholar] [CrossRef]
- Baliban, R.C.; Sakellari, D.; Li, Z.; Dimaggio, P.A.; Garcia, B.A.; Floudas, C.A. Novel protein identification methods for biomarker discovery via a proteomic analysis of periodontally healthy and diseased gingival crevicular fluid samples. J. Clin. Periodontol. 2012, 39, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.G.; Jehmlich, N.; Murr, A.; Dhople, V.M.; Holtfreter, B.; Hammer, E.; Völker, U.; Kocher, T. Identification of periodontitis associated changes in the proteome of whole human saliva by mass spectrometric analysis. J. Clin. Periodontol. 2013, 40, 825–832. [Google Scholar] [CrossRef]
- Grant, M.M.; Creese, A.J.; Barr, G.; Ling, M.R.; Scott, A.E.; Matthews, J.B.; Grifftihs, H.R.; Cooper, H.J.; Chapple, I.L.C. Proteomic analysis of a noninvasive human model of acute inflammation and its resolution: The twenty-one day gingivitis model. J. Proteome Res. 2010, 9, 4732–4744. [Google Scholar] [CrossRef] [Green Version]
- Feghali, K.; Grenier, D. Priming effect of fibronectin fragments on the macrophage inflammatory response: Potential contribution to periodontitis. Inflammation 2012, 35, 1696–1705. [Google Scholar] [CrossRef]
- Lamont, R.J.; Hajishengallis, G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015, 21, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Hajishengallis, E.; Kajikawa, T.; Wang, B.; Yancopoulou, D.; Ricklin, D.; Lambris, J.D. Complement inhibition in pre-clinical models of periodontitis and prospects for clinical application. Semin. Immunol. 2016, 28, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: a defence system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Aslam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darveau, R.P.; Hajishengallis, G.; Curtis, M.A. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 2012, 91, 816–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef]
- Choi, E.Y.; Chavakis, E.; Czabanka, M.A.; Langer, H.F.; Fraemohs, L.; Economopoulou, M.; Kundu, R.K.; Orlandi, A.; Zheng, Y.Y.; Prieto, D.A.; et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 2008, 322, 1101–1104. [Google Scholar] [CrossRef] [Green Version]
- Eskan, M.A.; Jotwani, R.; Abe, T.; Chmelar, J.; Lim, J.H.; Liang, S.; Ciero, P.A.; Kraus, J.L.; Li, F.; Rauner, H.; et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 2012, 13, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Deas, D.E.; Mackey, S.A.; McDonnell, H.T. Systemic disease and periodontitis: Manifestations of neutrophil dysfunction. Periodontol. 2000 2003, 32, 82–104. [Google Scholar] [CrossRef]
- Shin, J.; Maekawa, T.; Abe, T.; Hajishengallis, E.; Hosur, K.; Pyaram, K.; Mitroulis, I.; Chavakis, T.; Hajishengallis, G. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci. Transl. Med. 2015, 7, 307ra155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khader, S.A. Restraining IL-17: Del-1 deals the blow. Nat. Immunol. 2012, 13, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Chavakis, T. DEL-1-Regulated Immune Plasticity and Inflammatory Disorders. Trends Mol. Med. 2019, 25, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Mendes, K.L.; De Farias Lelis, D.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Golub, L.M.; Lee, H.M. Periodontal therapeutics: Current host-modulation agents and future directions. Periodontol. 2000 2020, 82, 186–204. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, E.; Ikeda, Y.; Wang, Y.; Fine, N.; Sheikh, Z.; Viniegra, A.; Barzilay, O.; Ganss, B.; Tenenbaum, H.C.; Glogauer, M. Resveratrol derivative-rich melinjo seed extract induces healing in a murine model of established periodontitis. J. Periodontol. 2018, 89, 586–595. [Google Scholar] [CrossRef]
- Marchesan, J.T.; Girnary, M.S.; Moss, K.; Monaghan, E.T.; Egnatz, G.J.; Jiao, Y.; Zhang, S.; Beck, J.; Swanson, K.V. Role of inflammasomes in the pathogenesis of periodontal disease and therapeutics. Periodontol. 2000 2020, 82, 93–114. [Google Scholar] [CrossRef]
- Marchesan, J.; Girnary, M.S.; Jing, L.; Miao, M.Z.; Zhang, S.; Sun, L.; Morelli, T.; Schoenfisch, M.H.; Inohara, N.; Offenbacher, S.; et al. An experimental murine model to study periodontitis. Nat. Protoc. 2018, 13, 2247–2267. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dee, K.C.; Puleo D, A.; Bizios, R. An Introduction To Tissue-Biomaterial Interactions; John Wiley & Sons, Inc.: New York, NY, USA, 2003. [Google Scholar] [CrossRef]
- Perretti, M.; Leroy, X.; Bland, E.J.; Montero-Melendez, T. Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol. Sci. 2015, 36, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Lumelsky, N.L. Commentary: Engineering of Tissue Healing and Regeneration. Tissue Eng. 2007, 13, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Hasturk, H.; Kantarci, A.; Goguet-Surmenian, E.; Blackwood, A.; Andry, C.; Serhan, C.N.; Van Dyke, T.E. Resolvin E1 Regulates Inflammation at the Cellular and Tissue Level and Restores Tissue Homeostasis In Vivo. J. Immunol. 2007, 179, 7021–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasturk, H.; Kantarci, A.; Ohira, T.; Arita, M.; Ebrahimi, N.; Chiang, N.; Petasis, N.; Levy, B.; Serhan, C.; Van Dyke, T.E. RvE1 protects from local inflammation and osteoclastmediated bone destruction in periodontitis. FASEB J. 2006, 20, 401–403. [Google Scholar] [CrossRef]
- Jönsson, D.; Ramberg, P.; Demmer, R.T.; Kebschull, M.; Dahlén, G.; Papapanou, P.N. Gingival tissue transcriptomes in experimental gingivitis. J. Clin. Periodontol. 2011, 38, 599–611. [Google Scholar] [CrossRef]
- Sorsa, T.; Alassiri, S.; Grigoriadis, A.; Räisänen, I.T.; Pärnänen, P.; Nwhator, S.O.; Gieselmann, D.R.; Sekallari, D. Active MMP-8 (AMMP-8) as a grading and staging biomarker in the periodontitis classification. Diagnostics 2020, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Räisänen, I.T.; Heikkinen, A.M.; Pakbaznejad Esmaeili, E.; Tervahartiala, T.; Pajukanta, R.; Silbereisen, A.; Bostanci, N.; Sorsa, T. A point-of-care test of active matrix metalloproteinase-8 predicts triggering receptor expressed on myeloid cells-1 (TREM-1) levels in saliva. J. Periodontol. 2020, 91, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Gul, S.S.; Douglas, C.W.I.; Griffiths, G.S.; Rawlinson, A. A pilot study of active enzyme levels in gingival crevicular fluid of patients with chronic periodontal disease. J. Clin. Periodontol. 2016, 43, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Gul, S.S.; Griffiths, G.S.; Stafford, G.P.; Al-Zubidi, M.I.; Rawlinson, A.; Douglas, C.W.I. Investigation of a Novel Predictive Biomarker Profile for the Outcome of Periodontal Treatment. J. Periodontol. 2017, 88, 1135–1144. [Google Scholar] [CrossRef]
- Huang, W.; He, B.Y.; Shao, J.; Jia, X.-W.; Yuan, Y.D. Interleukin-1β rs1143627 polymorphism with susceptibility to periodontal disease. Oncotarget 2017, 8, 31406–31414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-C.G.; Lerner, U.H.; Teng, Y.-T.A. Cytokine responses against periodontal infection: protective and destructive roles. Periodontol. 2000 2010, 52, 163–206. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qasim, S.S.B.; Al-Otaibi, D.; Al-Jasser, R.; Gul, S.S.; Zafar, M.S. An Evidence-Based Update on the Molecular Mechanisms Underlying Periodontal Diseases. Int. J. Mol. Sci. 2020, 21, 3829. https://doi.org/10.3390/ijms21113829
Qasim SSB, Al-Otaibi D, Al-Jasser R, Gul SS, Zafar MS. An Evidence-Based Update on the Molecular Mechanisms Underlying Periodontal Diseases. International Journal of Molecular Sciences. 2020; 21(11):3829. https://doi.org/10.3390/ijms21113829
Chicago/Turabian StyleQasim, Syed Saad B., Dalal Al-Otaibi, Reham Al-Jasser, Sarhang S. Gul, and Muhammad Sohail Zafar. 2020. "An Evidence-Based Update on the Molecular Mechanisms Underlying Periodontal Diseases" International Journal of Molecular Sciences 21, no. 11: 3829. https://doi.org/10.3390/ijms21113829
APA StyleQasim, S. S. B., Al-Otaibi, D., Al-Jasser, R., Gul, S. S., & Zafar, M. S. (2020). An Evidence-Based Update on the Molecular Mechanisms Underlying Periodontal Diseases. International Journal of Molecular Sciences, 21(11), 3829. https://doi.org/10.3390/ijms21113829