Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe


 ,
, 
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
Mitochondria: From Structure to Metabolic Pathways
2. A General Overview of Respiratory Chain Dysfunctions and Apoptotic Dysregulations
2.1. Disease-Associated Defects in Oxidative Phosphorylation
2.2. Apoptosis Dysregulation Increases the Propensity to Malignant Transformation
3. Mitochondria in the Hematopoietic Stem Cells: ADormant Galaxy
3.1. Low Energy Requirement Defines Hematopoietic Stem Cells
3.2. Low Free Radicals Amount: A Peculiar Feature of Hematopoietic Stem Cells
3.3. Mitochondrial Dynamics Defines Hematopoietic Stem Cells
4. Leukemia Stem Cells Mitochondria: A Vulnerable Dormant Galaxy
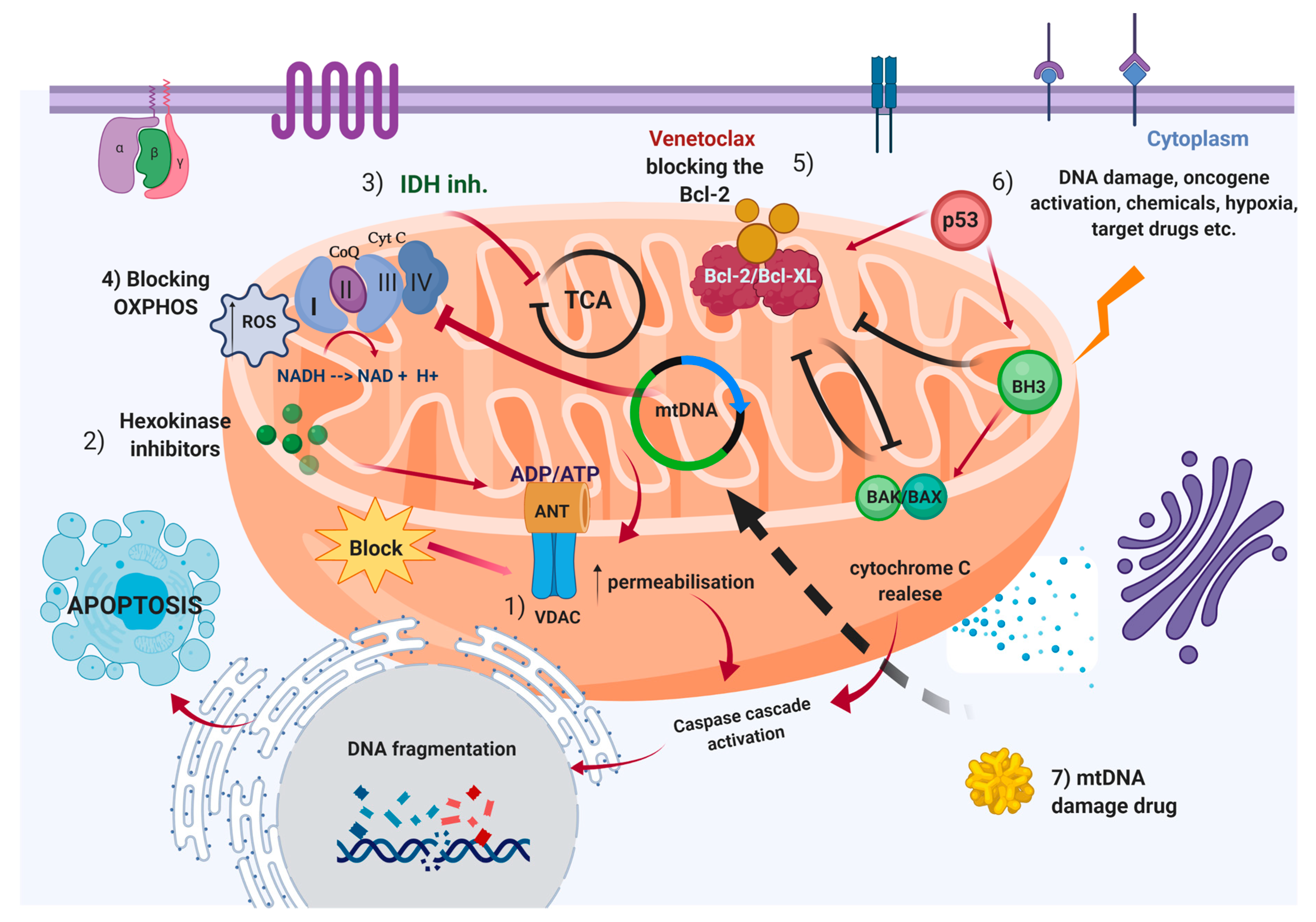
5. Mitochondria as Therapeutic “Hot-Spot” in Hematological Malignancies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VDAC | Voltage-dependent anion channel |
| MAM | Mitochondria-associated membranes |
| IMM | Inner mitochondrial membrane |
| RC | Respiratory chain |
| OMM | Outer mitochondrial membrane |
| ETC | Electron transport chain |
| ROS | Reactive oxygen species |
| TCGA | Cancer genome atlas |
| MDS | Myelodysplastic syndromes |
| AML | Acute myeloid leukemia |
| IDH | Isocitrate dehydrogenase |
| α-KG | α-ketoglutaric acid |
| CN-AML | Normal cytogenetics AML |
| COX | Cytochrome c oxidase |
| CLL | Chronic lymphocytic leukemia |
| OMM | Outer mitochondriamembrane |
| HSC | Hematopoietic stem cells |
| OXPHOS | Oxidative phosphorylation program |
| PDK | Pyruvate dehydrogenase kinase |
| HIF-1 | Hypoxia inducible factor 1 |
| LDHA | Lactate dehydrogenase A |
| HK1, HK2 | Hexokinase 1 and 2 |
| ALD-A, ALD-C | Aldolase A and C |
| ENOalpha | Enolase alpha |
| PGK1 | Phosphoglycerate kinase 1 |
| NFR2 | Nuclear factor erythroid 2-related factor 2 |
| SODs | Superoxide dismutase genes |
| FoxO3 | Forkhead box O3 |
| PTEN | Phosphatase and tensin homolog |
| AMPK | 5′ AMP-activated protein kinase |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| PIP2 | Fosfatidilinositolo 4,5-bisfosfato |
| PIP3 | Phosphoinositide-3,4,5-trisphosphate |
| ATM | Ataxia telangiectasia mutated |
| BID | BH3-interacting domain death agonist |
| Drp1 | Dynamin-related protein 1 |
| Fis1 | Mitochondrial fission 1 protein |
| OPA1 | Mitochondrial dynamin like GTPase |
| Atg | Autophagy-related genes |
| PINK1 | PTEN-induced putative kinase 1 |
| TSC1/2 | Tuberous sclerosis 1/2 |
| PPAR δ | Peroxisome proliferator-activated receptor delta |
| CSC | Cancer stem cell |
| FAO | Fatty amino acid metabolism |
| GLS | Enzyme glutaminase |
| GSH | Glutathione |
| ABT-199 | Venetoclax |
| GPX3 | Glutathione peroxidase 3 |
| ANT | Adenine nucleotide translocator |
References
- Martin, W.F.; Müller, M. Origin of Mitochondria and Hydrogenosomes; Springer: New York, NY, USA, 2007. [Google Scholar]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.G.E.; Zomorodipour, A.; Andersson, J.O.; Sicheritz-Ponten, T.; Alsmark, U.C.M.; Podowski, R.M.; Näslund, A.K.; Eriksson, A.-S.; Winkler, H.H.; Kurland, C.G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 1998, 396, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef]
- Gomes, L.C.; di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation, and sustain cell viability. Nature 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Nat. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; E Haigh, S.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Murley, A.; Nunnari, J. The Emerging Network of Mitochondria-Organelle Contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M.; Blachly-Dyson, E.; Forte, M. VDAC, a Channel in the Outer Mitochondrial Membrane. Adv. Struct. Saf. Stud. 1996, 4, 169–202. [Google Scholar] [CrossRef]
- Acín-Pérez, R.; Silva, P.F.; Peleato, M.L.; Pérez-Martos, A.; Enríquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Cogliati, S.; Enríquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pr. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Bradley, R.M.; Stark, K.D.; Duncan, R.E. Influence of tissue, diet, and enzymatic remodeling on cardiolipin fatty acyl profile. Mol. Nutr. Food Res. 2016, 60, 1804–1818. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nature 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Dudek, J.; Cheng, I.-F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bömeke, K.; Hübscher, D.; Vukotic, M.; Wanders, R.; Rehling, P.; Guan, K. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef] [Green Version]
- Nisoli, E.; Cozzi, V.; Carruba, M.O. Amino Acids and Mitochondrial Biogenesis. Am. J. Cardiol. 2008, 101, 22–25. [Google Scholar] [CrossRef]
- Guda, P.; Guda, C.; Subramaniam, S. Reconstruction of Pathways Associated with Amino Acid Metabolism in Human Mitochondria. Genom. Proteom. Bioinform. 2007, 5, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, Z.; Khan, A.; Zheng, H.; Yuan, C.; Jiang, H. Advances in drug therapy for mitochondrial diseases. Ann. Transl. Med. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cytopathies. Cell Calcium 2016, 60, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-R.; Shi, Y. Chloramphenicol Induces Abnormal Differentiation, and Inhibits Apoptosis in Activated T Cells. Cancer Res. 2008, 68, 4875–4881. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-H.; Cheng, Y.-W.; Liao, P.-L.; Yang, Y.-T.; Kang, J.-J. Chloramphenicol causes mitochondrial stress, decreases ATP biosynthesis, induces matrix metalloproteinase-13 expression, and solid-tumor cell invasion. Toxicol. Sci. 2010, 116, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-H.; Tzeng, S.-L.; Cheng, Y.-W.; Kang, J.-J. Chloramphenicol-induced Mitochondrial Stress Increases p21 Expression and Prevents Cell Apoptosis through a p21-dependent Pathway. J. Biol. Chem. 2005, 280, 26193–26199. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Ylikallio, E.; Suomalainen, A. Mechanisms of mitochondrial diseases. Ann. Med. 2011, 44, 41–59. [Google Scholar] [CrossRef]
- Wu, S.; Akhtari, M.; Alachkar, H. Characterization of mutations in the mitochondrial encoded electron transport chain complexes in acute myeloid leukemia. Sci. Rep. 2018, 8, 13301. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Fato, R.; Genova, M.L.; Bergamini, C.; Bianchi, C.; Biondi, A. Mitochondrial Complex I: Structural and functional aspects. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1406–1420. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2011, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahid, M.; Pourrut, B.; Dumat, C.; Nadeem, M.; Aslam, M.; Pinelli, E. Heavy-Metal-Induced Reactive Oxygen Species: Phytotoxicity and Physicochemical Changes in Plants. Rev. Env. Contam. Toxicol. 2014, 232, 1–44. [Google Scholar] [CrossRef]
- de Giusti, V.C.; Caldiz, C.I.; Ennis, I.; Pérez, N.G.; Cingolani, H.E.; Aiello, E.A. Mitochondrial reactive oxygen species (ROS) as signaling molecules of intracellular pathways triggered by the cardiac renin-angiotensin II-aldosterone system (RAAS). Front. Physiol. 2013, 4, 126. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, S. Modulation of Reactive Oxygen Species in Health and Disease. Antioxidants 2019, 8, 513. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G. The Mitochondrial Production of Reactive Oxygen Species: Mechanisms and Implications in Human Pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free. Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Lenaz, G.; Baracca, A.; Fato, R.; Genova, M.L.; Solaini, G. New Insights into Structure and Function of Mitochondria and Their Role in Aging and Disease. Antioxidants Redox Signal. 2006, 8, 417–437. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Pätsi, J.; Kervinen, M.; Finel, M.; Hassinen, I. Leber hereditary optic neuropathy mutations in the ND6 subunit of mitochondrial complex I affect ubiquinone reduction kinetics in a bacterial model of the enzyme. Biochem. J. 2008, 409, 129–137. [Google Scholar] [CrossRef]
- Gatt, A.P.; Duncan, O.F.; Attems, J.; Francis, P.T.; Ballard, C.G.; Bateman, J.M. Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex i deficiency. Movement disorders: Official. J. Mov. Dis. Soc. 2016, 31, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2018, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Bunke, T.; Thol, F.; Márkus, B.; Wagner, K.; Göhring, G.; Schlegelberger, B.; Heil, G.; Reuter, C.W.M.; Püllmann, K.; et al. Prognostic implications and molecular associations of NADH dehydrogenase subunit 4 (ND4) mutations in acute myeloid leukemia. Leukemia 2011, 26, 289–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Owain, M.; Colak, D.; AlBakheet, A.; Al-Younes, B.; Al-Humaidi, Z.; Al-Sayed, M.; Al-Hindi, H.; Al-Sugair, A.; Al-Muhaideb, A.; Rahbeeni, Z.; et al. Clinical and biochemical features associated with BCS1L mutation. J. Inherit. Metab. Dis. 2012, 36, 813–820. [Google Scholar] [CrossRef]
- Barletta, J.A.; Hornick, J.L. Succinate Dehydrogenase-deficient Tumors. Adv. Anat. Pathol. 2012, 19, 193–203. [Google Scholar] [CrossRef]
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; A Oldenburg, R.; A De Bruyn, E.M.C.; Sleddens, H.F.B.M.; Derkx, P.; Rivière, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Leigh and Leigh-Like Syndrome in Children and Adults. Pediatr. Neurol. 2008, 39, 223–235. [Google Scholar] [CrossRef]
- Skillings, E.A.; Morton, A.J. Delayed onset and reduced cognitive deficits through pre-conditioning with 3-nitropropionic acid is dependent on sex and cag repeat length in the r6/2 mouse model of Huntington’s disease. J. Huntingtons Dis. 2016, 5, 19–32. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019, 134, 389–394. [Google Scholar] [CrossRef]
- Xia, D.; Esser, L.; Tang, W.-K.; Zhou, F.; Zhou, Y.; Yu, L.; Yu, C.-A. Structural analysis of cytochrome bc1 complexes: Implications to the mechanism of function. Biochim. Biophys. Acta Bioenerg. 2012, 1827, 1278–1294. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free. Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martínez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Vizarra, E.; Zeviani, M. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front. Genet. 2015, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Signes, A.; Fernández-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I–V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P.; Marshall, D.C.; Cooper, C.; Wilson, M.T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013, 41, 1312–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, J.D. Chromosome studies in the non-hodgkin’s lymphomas: The role of the 14;18 translocation. J. Clin. Oncol. 1988, 6, 919–925. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Silkjaer, T.; Nyvold, C.G.; Juhl-Christensen, C.; Hokland, P.; Nørgaard, J.M. Mitochondrial cytochromecoxidase subunit II variations predict adverse prognosis in cytogenetically normal acute myeloid leukaemia. Eur. J. Haematol. 2013, 91, 295–303. [Google Scholar] [CrossRef]
- Silkjaer, T.; Nørgaard, J.M.; Aggerholm, A.; Ebbesen, L.H.; Kjeldsen, E.; Hokland, P.; Nyvold, C.G. Characterization and prognostic significance of mitochondrial DNA variations in acute myeloid leukemia. Eur. J. Haematol. 2013, 90, 385–396. [Google Scholar] [CrossRef]
- Kluckova, K.; Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Neuzil, J. Mitochondrial complex II, a novel target for anti-cancer agents. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Amaya, M.L.; Pollyea, D.A. Targeting theIDH2Pathway in Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 4931–4936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcucci, G.; Haferlach, T.; Döhner, H. Molecular Genetics of Adult Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. J. Clin. Oncol. 2011, 29, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Fathi, A.T.; Dinardo, C.D.; A Pollyea, D.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2016, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; Van Noorden, C.J.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta Bioenerg. 2014, 1846, 326–341. [Google Scholar] [CrossRef]
- Chan, S.M.; Majeti, R. Role of dnmt3a, tet2, and idh1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Letai, A. Apoptosis and Cancer. Annu. Rev. Cancer Biol. 2017, 1, 275–294. [Google Scholar] [CrossRef]
- Hanahan, D.; A Weinberg, R. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- McLangemeijer, S.; Mariani, N.; Knops, R.; Gilissen, C.; Woestenenk, R.; De Witte, T.; Huls, G.; Van Der Reijden, B.; Jansen, J.H. Apoptosis-Related Gene Expression Profiling in Hematopoietic Cell Fractions of MDS Patients. PLoS ONE 2016, 11, e0165582. [Google Scholar] [CrossRef]
- McBride, A.; Houtmann, S.; Wilde, L.; Vigil, C.; Eischen, C.M.; Kasner, M.; Palmisiano, N.D. The Role of Inhibition of Apoptosis in Acute Leukemias and Myelodysplastic Syndrome. Front. Oncol. 2019, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, S.; Rowley, J.D.; Variakojis, D.; Golomb, H.M. Chromosome abnormalities in poorly differentiated lymphocytic lymphoma. Cancer Res. 1979, 39, 3119–3128. [Google Scholar] [PubMed]
- Tsujimoto, Y.; Finger, L.; Yunis, J.; Nowell, P.; Croce, C. Cloning of the chromosome breakpoint of neoplastic B cells with the t (14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Bcl-2 family proteins: Regulators of apoptosis and chemoresistance in hematologic malignancies. Semin. Hematol. 1997, 34, 9–19. [Google Scholar] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Zhou, J.-D.; Zhang, T.-J.; Xu, Z.-J.; Gu, Y.; Ma, J.-C.; Li, X.-X.; Guo, H.; Wen, X.-M.; Zhang, W.; Yang, L.; et al. BCL2 overexpression: Clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019, 14, 68. [Google Scholar] [CrossRef]
- Boudard, D.; Vasselon, C.; Berthéas, M.-F.; Jaubert, J.; Mounier, C.; Reynaud, J.; Viallet, A.; Chautard, S.; Guyotat, D.; Campos, L. Expression and prognostic significance of Bcl-2 family proteins in myelodysplastic syndromes. Am. J. Hematol. 2002, 70, 115–125. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef] [Green Version]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, N.D. The Release of Cytochrome c from Mitochondria: A Primary Site for Bcl-2 Regulation of Apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Korsmeyer, S.J.; Schlesinger, P. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nature 2000, 2, 553–555. [Google Scholar] [CrossRef]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic bax and bak: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, J.M.; Westphal, D.; Dewson, G.; Robin, A.; Uren, R.T.; Bartolo, R.; Thompson, G.V.; Colman, P.M.; Kluck, R.M.; Czabotar, P.E. Bak Core and Latch Domains Separate during Activation, and Freed Core Domains Form Symmetric Homodimers. Mol. Cell 2014, 55, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nature 2006, 8, 1348–1358. [Google Scholar] [CrossRef]
- Zhang, Z.; Subramaniam, S.; Kale, J.; Liao, C.; Huang, B.; Brahmbhatt, H.; Condon, S.; Lapolla, S.M.; A Hays, F.; Ding, J.; et al. BH 3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2015, 35, 208–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Mooers, B.; Zhang, Z.; Kale, J.; Falcone, M.; McNichol, J.; Huang, B.; Zhang, X.C.; Xing, C.; Andrews, D.W.; et al. After Embedding in Membranes Antiapoptotic Bcl-XL Protein Binds Both Bcl-2 Homology Region 3 and Helix 1 of Proapoptotic Bax Protein to Inhibit Apoptotic Mitochondrial Permeabilization. J. Biol. Chem. 2014, 289, 11873–11896. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, F.; Wiedenmann, C.; Schalk, E.; Becker, D.; Funk, K.; Scholz-Kreisel, P.; Todt, F.; Wolleschak, D.; Döhner, K.; Marquardt, J.U.; et al. Mitochondrial BAX Determines the Predisposition to Apoptosis in Human AML. Clin. Cancer Res. 2017, 23, 4805–4816. [Google Scholar] [CrossRef] [Green Version]
- Reyna, D.E.; Garner, T.P.; Lopez, A.; Kopp, F.; Choudhary, G.S.; Sridharan, A.; Narayanagari, S.-R.; Mitchell, K.; Dong, B.; Bartholdy, B.A.; et al. Direct Activation of BAX by BTSA1 Overcomes Apoptosis Resistance in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 490–505. [Google Scholar] [CrossRef] [Green Version]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Heiden, M.G.V.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [Green Version]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ocampo, A.; Belmonte, J.C.I. Cellular Metabolism, and Induced Pluripotency. Cell 2016, 166, 1371–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Lopez, S.; Lerner, R.G.; Petritsch, C.K. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell. Mol. Life Sci. 2013, 71, 575–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, J.G.; Gardner, D.K.; Harvey, A. Pluripotent Stem Cell Metabolism and Mitochondria: Beyond ATP. Stem Cells Int. 2017, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Dev. 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two-decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Chunzhu, T.; Wei, L. Ldha is a feedback activator of hypoxia inducible factor 1-alpha in ovarian cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 10437–10443. [Google Scholar]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation ofglut1mRNA by Hypoxia-inducible Factor-1. J. Biol. Chem. 2000, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Li, Q.; Tang, F.; Puchowitz, M.A.; Fujioka, H.; Dunwoodie, S.L.; Danielpour, D.; Yang, Y.-C. Cited2 Is Required for the Maintenance of Glycolytic Metabolism in Adult Hematopoietic Stem Cells. Stem Cells Dev. 2013, 23, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Unnisa, Z.; Clark, J.P.; Roychoudhury, J.; Thomas, E.; Tessarollo, L.; Copeland, N.G.; Jenkins, N.A.; Grimes, L.; Kumar, A.R. Meis1 preserves hematopoietic stem cells in mice by limiting oxidative stress. Blood 2012, 120, 4973–4981. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Carracedo, A.; Weiss, O.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Bioenerg. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Kansanen, E.; Linna-Kuosmanen, S.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.-H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs Are Lineage-Restricted Redundant Tumor Suppressors and Regulate Endothelial Cell Homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.P.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-C.; Jeon, S.-M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs Inhibit mTORC1 and Activate Akt by Inducing the Expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kops, G.J.P.L.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.A.; Coffer, P.J.; Huang, T.-T.; Bos, J.L.; Medema, R.H.; Burgering, B. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Tan, W.-Q.; Wang, K.; Lv, D.-Y.; Li, P. Foxo3a Inhibits Cardiomyocyte Hypertrophy through Transactivating Catalase. J. Biol. Chem. 2008, 283, 29730–29739. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, J.; Kapur, R. Regulation of Hematopoietic Stem Cell Self-Renewal and Leukemia Maintenance by the PI3K-mTORC1 Pathway. Curr. Stem Cell Rep. 2016, 2, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jin, H.P.; Woojin, J.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [Green Version]
- Bankoglu, E.E.; Tschopp, O.; Schmitt, J.; Burkard, P.; Jahn, D.; Geier, A.; Stopper, H. Role of PTEN in Oxidative Stress and DNA Damage in the Liver of Whole-Body Pten Haplodeficient Mice. PLoS ONE 2016, 11, e0166956. [Google Scholar] [CrossRef]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The Energy Sensor AMP-activated Protein Kinase Directly Regulates the Mammalian FOXO3 Transcription Factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Zhang, C.; Yu, B.; Chen, B.; Liu, Z.; Hou, C.; Wang, F.; Shen, H.; Chen, Z. Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am. J. Cancer Res. 2017, 7, 1407–1422. [Google Scholar] [PubMed]
- Feige, J.N.; Auwerx, J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr. Opin. Cell Biol. 2008, 20, 303–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Bhatia, R. Role of SIRT1 in the growth and regulation of normal hematopoietic and leukemia stem cells. Curr. Opin. Hematol. 2015, 22, 324–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, K.D.; Palaniappan, V.V.; Espinosa, J.M. ATM regulates cell fate choice upon p53 activation by modulating mitochondrial turnover and ROS levels. Cell Cycle 2014, 14, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Valentin-Vega, Y.A.; MacLean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Gross, A.; Zaltsman, Y.; Maryanovich, M. The ATM–BID pathway plays a critical role in the DNA damage response by regulating mitochondria metabolism. Cell Death Differ. 2015, 23, 182. [Google Scholar] [CrossRef] [Green Version]
- Çam, H.; Easton, J.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Chow, H.-M.; Cheng, A.; Song, X.; Swerdel, M.R.; Hartlova, A.; Herrup, K. ATM is activated by ATP depletion and modulates mitochondrial function through NRF1. J. Cell Biol. 2019, 218, 909–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinge, A.; He, J.; Bartram, J.; Javier, J.; Xu, J.; Fjellman, E.; Sesaki, H.; Li, T.; Yu, J.; Wunderlich, M.; et al. Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 2020, 26, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, L.P.; Chandel, N.S. HSC Fate Is Tethered to Mitochondria. Cell Stem Cell 2016, 18, 303–304. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, L.L.; De Almeida, M.J.; Corrigan, D.J.; Mumau, M.; Snoeck, H.-W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 2016, 529, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef]
- Koschade, S.E.; Brandts, C.H. Selective Autophagy in Normal and Malignant Hematopoiesis. J. Mol. Biol. 2020, 432, 261–282. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; De La Cruz-Ojeda, P.; De La Mata, M.; Cotán, D.; Oropesa-Ávila, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.; Kundu, M. Mitophagy in hematopoietic stem cells. Autophagy 2013, 9, 1737–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Youle, R.J. Targeting Mitochondrial Dysfunction: Role for PINK1 and Parkin in Mitochondrial Quality Control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef]
- Nakada, D.; Saunders, T.L.; Morrison, S.J. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 2010, 468, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.-C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- Dick, J.E. Acute myeloid leukemia stem cells. Anna. New York Acad. Sci. 2005, 1044, 1–5. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nature reviews. Cancer 2004, 4, 891–899. [Google Scholar]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagust, P.; de Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattes, K.; Vellenga, E.; Schepers, H. Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: A potential window for leukemia therapy. Crit. Rev. Oncol. 2019, 144, 102814. [Google Scholar] [CrossRef]
- Ito, K.; Ito, K. Hematopoietic stem cell fate through metabolic control. Experim. Hematol. 2018, 64, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Karigane, D.; Takubo, K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int. J. Hematol. 2017, 106, 18–26. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; de Gregori, J.; et al. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; Culp-Hill, R.; Dalessandro, A.; Krug, A.; Goosman, M.; Pei, S.; A Pollyea, D.; Jordan, C.T. Inhibition of Fatty Acid Metabolism Re-Sensitizes Resistant Leukemia Stem Cells to Venetoclax with Azacitidine. Blood 2019, 134, 1272. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabe, Y.; Konopleva, M.; Andreeff, M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front. Oncol. 2020, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, N.; Poulain, L.; Birsen, R.; Tamburini, J.; Bouscary, D. Rationale for Targeting Deregulated Metabolic Pathways as a Therapeutic Strategy in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Herault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Chagraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; An Andrade-Navarro, M.; Hébert, J.; et al. A role for GPx3 in activity of normal and leukemia stem cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Huang, X.; Lee, M.R.; A Lee, S.; Broxmeyer, H.E. Antagonism of PPAR-γ signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat. Med. 2018, 24, 360–367. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [Green Version]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [Green Version]
- Shafat, M.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Lee, E.A.; Angka, L.; Rota, S.-G.; Hanlon, T.; Mitchell, A.; Hurren, R.; Wang, X.-M.; Gronda, M.; Boyaci, E.; Bojko, B.; et al. Targeting Mitochondria with Avocatin B Induces Selective Leukemia Cell Death. Cancer Res. 2015, 75, 2478–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Cancelas, J.A. Gap Junctions in the Bone Marrow Lympho-Hematopoietic Stem Cell Niche, Leukemia Progression, and Chemoresistance. Int. J. Mol. Sci. 2020, 21, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presley, C.A.; Lee, A.W.; Kastl, B.; Igbinosa, I.; Yamada, Y.; Fishman, G.I.; Gutstein, D.E.; Cancelas, J.A. Bone Marrow Connexin-43 Expression Is Critical for Hematopoietic Regeneration After Chemotherapy. Cell Commun. Adhes. 2005, 12, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Nat. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Best, G.; Xia, W.; Peters, L.; Wong, K.; Ward, C.; Greenwood, M. Expression of Intracellular Reactive Oxygen Species in Hematopoietic Stem Cells Correlates with Time to Neutrophil and Platelet Engraftment in Patients Undergoing Autologous Bone Marrow Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 1997–2002. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef] [Green Version]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [Green Version]
- Picou, F.; Vignon, C.; Debeissat, C.; Lachot, S.; Kosmider, O.; Gallay, N.; Foucault, A.; Estienne, M.-H.; Ravalet, N.; Béné, M.C.; et al. Bone marrow oxidative stress and specific antioxidant signatures in myelodysplastic syndromes. Blood Adv. 2019, 3, 4271–4279. [Google Scholar] [CrossRef] [Green Version]
- Asai, T.; Liu, Y.; Bae, N.; Nimer, S.D. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J. Cell. Physiol. 2011, 226, 2215–2221. [Google Scholar] [CrossRef] [Green Version]
- Voltan, R.; Secchiero, P.; Corallini, F.; Zauli, G. Selective induction of tp53i3/p53-inducible gene 3 (pig3) in myeloid leukemia cells, but not in normal cells, by nutlin-3. Mol. Carcinog. 2014, 53, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Hogdal, L.J.; Letai, A. BCL-2 inhibition: Stemming the tide of myeloid malignancies. Cell Stem Cell 2013, 12, 269–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.S.; Kim, S.Y.; Fein, J.A.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Leni, Z.; Parakkal, G.; Arcaro, A. Emerging Metabolic Targets in the Therapy of Hematological Malignancies. BioMed Res. Int. 2013, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Roboz, G.J.; Dinardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.L.; Trinh, D.L.; Ries, R.E.; Wang, J.; Gerbing, R.B.; Ma, Y.; Topham, J.; Hughes, M.; Pleasance, E.; Mungall, A.J.; et al. MicroRNA Expression-Based Model Indicates Event-Free Survival in Pediatric Acute Myeloid Leukemia. J. Clin. Oncol. 2017, 35, 3964–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. New Engl. J. Med. 2015, 374, 311–322. [Google Scholar] [CrossRef]
- Deng, J.; Carlson, N.; Takeyama, K.; Cin, P.D.; Shipp, M.; Letai, A. BH3 Profiling Identifies Three Distinct Classes of Apoptotic Blocks to Predict Response to ABT-737 and Conventional Chemotherapeutic Agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Campos, E.D.V.; Pinto, R. Targeted therapy with a selective BCL-2 inhibitor in older patients with acute myeloid leukemia. Hematol. Transfus. Cell Ther. 2019, 41, 169–177. [Google Scholar] [CrossRef]
- Han, L.; Zhang, Q.; Dail, M.; Shi, C.; Cavazos, A.; Ruvolo, V.R.; Zhao, Y.; Kim, E.; Rahmani, M.; Mak, D.H.; et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematology 2019, 105, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Bose, P.; Gandhi, V.V.; Konopleva, M.Y. Pathways and mechanisms of venetoclax resistance. Leuk. Lymphoma 2017, 58, 1–17. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Aldoss, I.; Yang, D.; Aribi, A.; Ali, H.; Sandhu, K.; Al Malki, M.M.; Mei, M.; Salhotra, A.; Khaled, S.; Nakamura, R.; et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematology 2018, 103, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Lanni, C.; Govoni, S.; Uberti, D.; D’Orazi, G.; Racchi, M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: Is HIPK2 the link? Aging 2010, 2, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: P53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, T.; Lozano, G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Faderl, S.; Kantarjian, H.M.; Estey, E.; Manshouri, T.; Chan, C.Y.; Rahman Elsaied, A.; Kornblau, S.M.; Cortes, J.; Thomas, D.A.; Pierce, S.; et al. The prognostic significance of p16(ink4a)/p14(arf) locus deletion and mdm-2 protein expression in adult acute myelogenous leukemia. Cancer 2000, 89, 1976–1982. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2017, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, V.P.; Initiative, A.P.C.G.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; MacLean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panuzzo, C.; Jovanovski, A.; Pergolizzi, B.; Pironi, L.; Stanga, S.; Fava, C.; Cilloni, D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int. J. Mol. Sci. 2020, 21, 3928. https://doi.org/10.3390/ijms21113928
Panuzzo C, Jovanovski A, Pergolizzi B, Pironi L, Stanga S, Fava C, Cilloni D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. International Journal of Molecular Sciences. 2020; 21(11):3928. https://doi.org/10.3390/ijms21113928
Chicago/Turabian StylePanuzzo, Cristina, Aleksandar Jovanovski, Barbara Pergolizzi, Lucrezia Pironi, Serena Stanga, Carmen Fava, and Daniela Cilloni. 2020. "Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe" International Journal of Molecular Sciences 21, no. 11: 3928. https://doi.org/10.3390/ijms21113928
APA StylePanuzzo, C., Jovanovski, A., Pergolizzi, B., Pironi, L., Stanga, S., Fava, C., & Cilloni, D. (2020). Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. International Journal of Molecular Sciences, 21(11), 3928. https://doi.org/10.3390/ijms21113928