Fractionation of Enriched Phosphopeptides Using pH/Acetonitrile-Gradient-Reversed-Phase Microcolumn Separation in Combination with LC–MS/MS Analysis


Abstract
:1. Introduction
2. Results
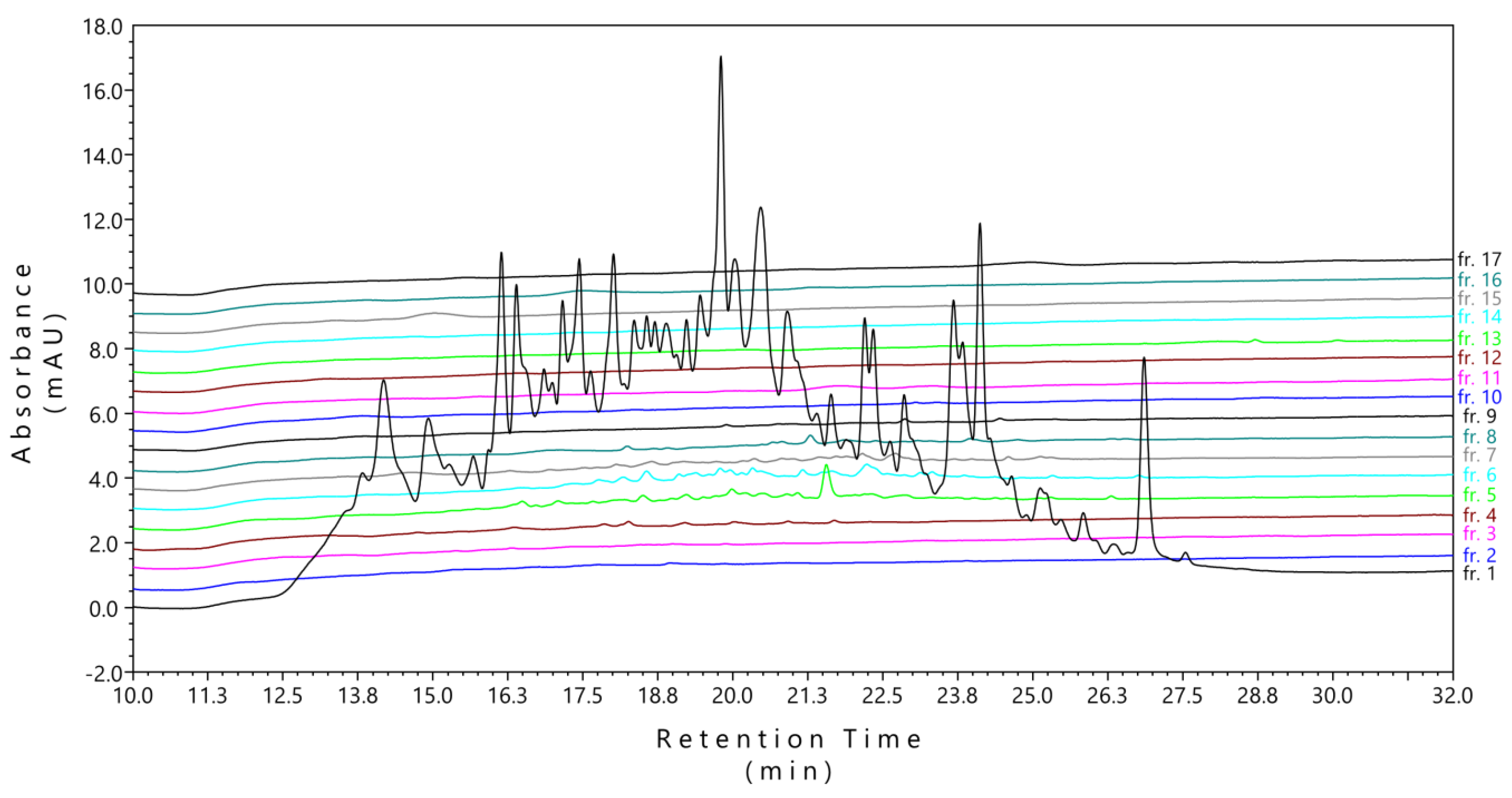
2.1. Conventional High-pH-RP Fractionation Was not Suitable for Phosphopeptides
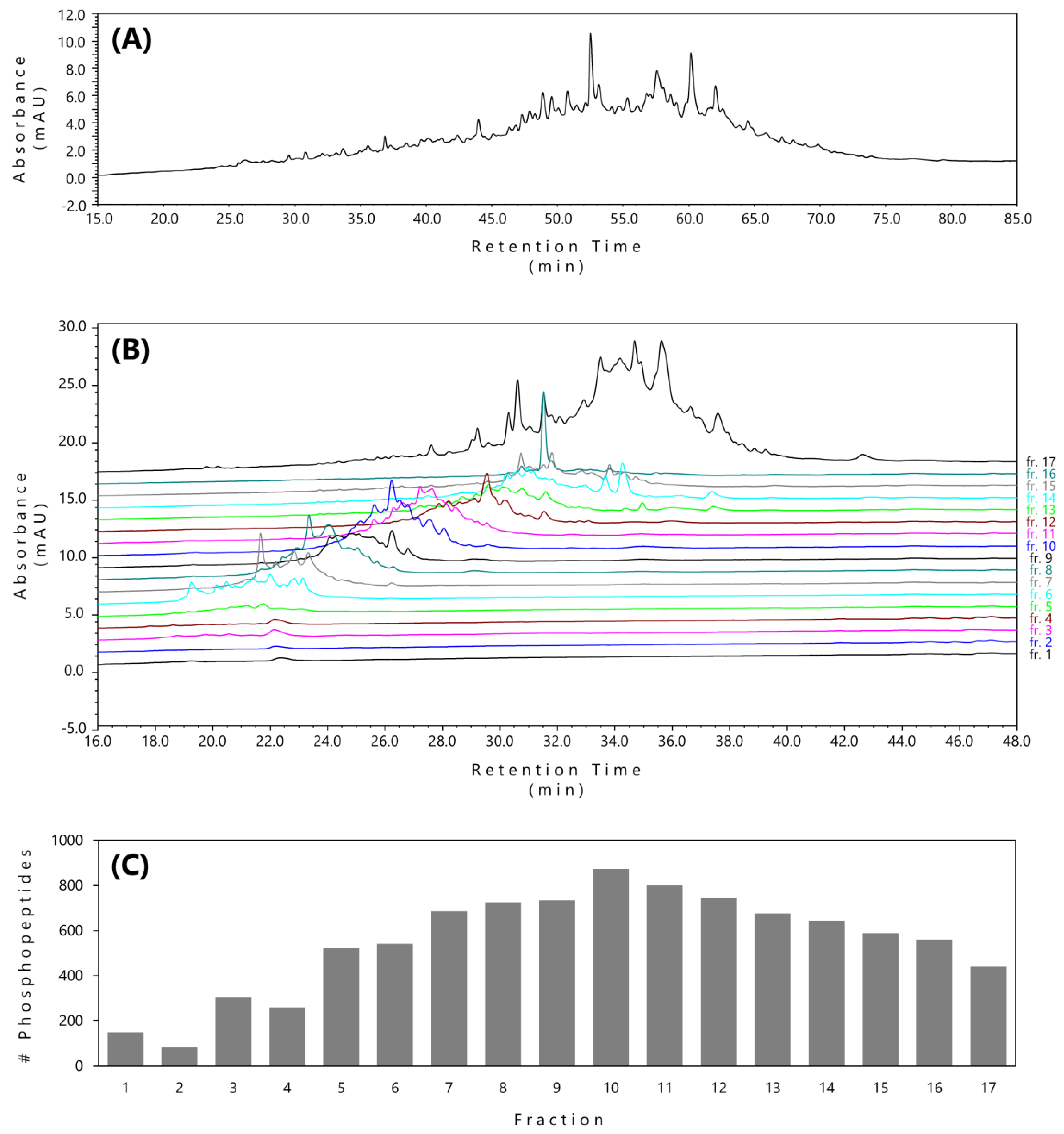
2.2. pH/ACN-Gradient-RP Procedure Provided Improved Phosphopeptide Fractionation
2.3. LC–MS/MS Analysis Confirmed a Significant Improvement in Phosphopeptide Fractionation
3. Discussion
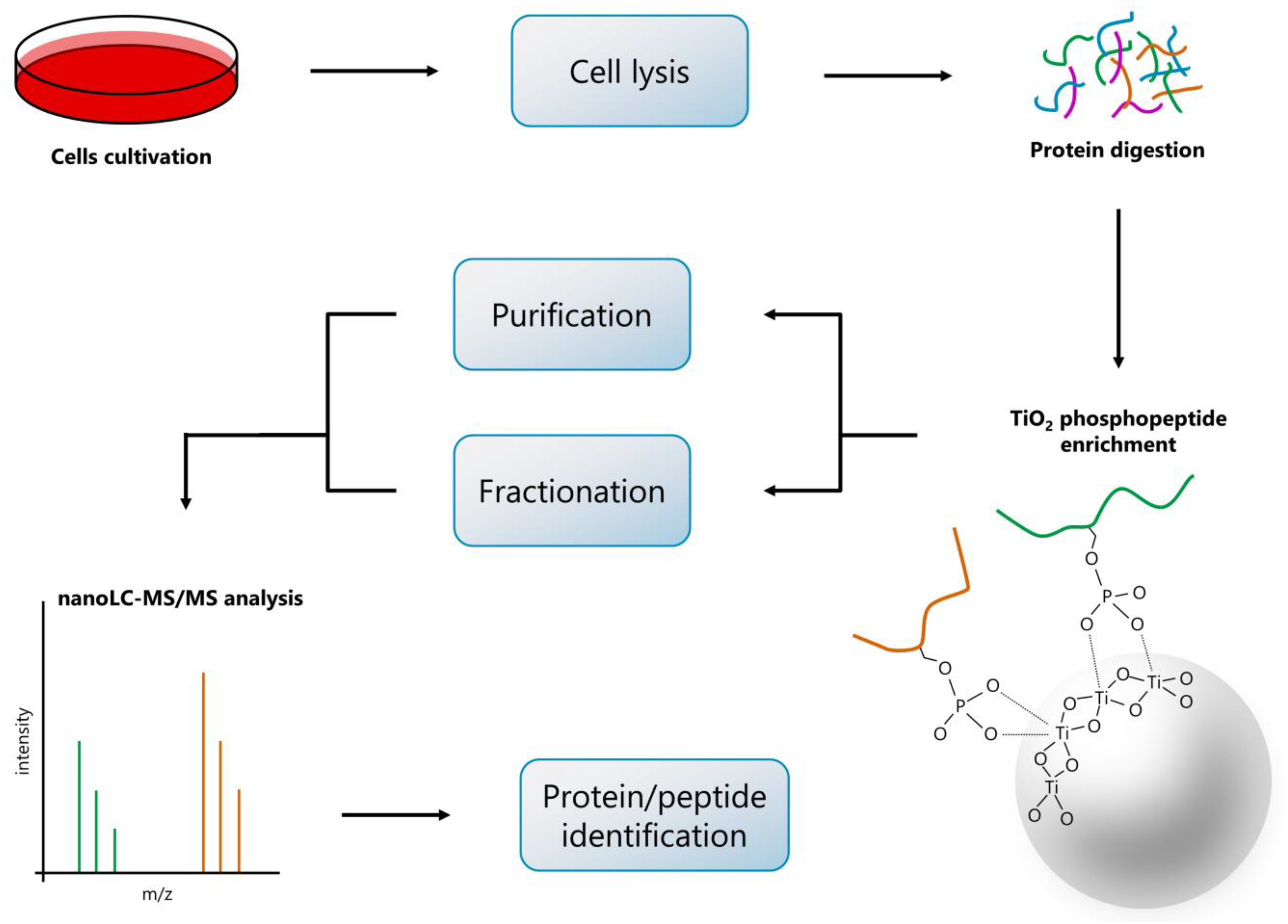
4. Materials and Methods
4.1. Cell Cultures and Cultivation
4.2. Cell Treatment and Recovery of Proteins
4.3. Enrichment for Phosphopeptides
4.4. Purification of Phosphopeptides
4.5. Fractionation of Phosphopeptides
4.6. Liquid Chromatography and Mass Spectrometry Analysis
4.7. Phosphoproteomic Data Processing
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlastaridis, P.; Kyriakidou, P.; Chaliotis, A.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. Gigascience 2017, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.X.; Gritsenko, M.A.; Smith, R.D.; Qian, W.-J. The current state of the art of quantitative phosphoproteomics and its applications to diabetes research. Expert Rev. Proteom. 2016, 13, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayya, V.; Han, D.K. Phosphoproteomics by Mass Spectrometry: insights, implications, applications, and limitations. Expert Rev. Proteom. 2009, 6, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Tichy, A.; Salovska, B.; Rehulka, P.; Klimentova, J.; Vavrova, J.; Stulik, J.; Hernychova, L. Phosphoproteomics: searching for a needle in a haystack. J. Proteom. 2011, 74, 2786–2797. [Google Scholar] [CrossRef]
- Fíla, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.D.; Reid, G.E.; Bruening, M.L. Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom. Rev. 2010, 29, 29–54. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Larsen, M.R. The use of titanium dioxide for selective enrichment of phosphorylated peptides. Methods Mol. Biol. 2016, 1355, 135–146. [Google Scholar] [CrossRef]
- Pinkse, M.W.H.; Uitto, P.M.; Hilhorst, M.J.; Ooms, B.; Heck, A.J.R. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 2004, 76, 3935–3943. [Google Scholar] [CrossRef]
- Ruprecht, B.; Koch, H.; Medard, G.; Mundt, M.; Kuster, B.; Lemeer, S. Comprehensive and reproducible phosphopeptide enrichment using iron immobilized metal ion affinity chromatography (Fe-IMAC) columns. Mol. Cell. Proteom. 2015, 14, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenselau, C. A review of quantitative methods for proteomic studies. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 855, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Nita-Lazar, A.; Saito-Benz, H.; White, F.M. Quantitative phosphoproteomics by mass spectrometry: past, present, and future. Proteomics 2008, 8, 4433–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deracinois, B.; Flahaut, C.; Duban-Deweer, S.; Karamanos, Y. Comparative and quantitative global proteomics approaches: an overview. Proteomes 2013, 1, 180–218. [Google Scholar] [CrossRef] [Green Version]
- Manadas, B.; Mendes, V.M.; English, J.; Dunn, M.J. Peptide fractionation in proteomics approaches. Expert Rev. Proteom. 2010, 7, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.; Wasinger, V.C. Protein and peptide fractionation, enrichment and depletion: tools for the complex proteome. Proteomics 2011, 11, 513–534. [Google Scholar] [CrossRef]
- Cao, Z.; Tang, H.-Y.; Wang, H.; Liu, Q.; Speicher, D.W. Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J. Proteome Res. 2012, 11, 3090–3100. [Google Scholar] [CrossRef] [Green Version]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28, 1694–1703. [Google Scholar] [CrossRef]
- Que, A.H.; Kahle, V.; Novotny, M.V. A microgradient elution system for capillary electrochromatography. J. Microcolumn Sep. 2000, 12, 1–5. [Google Scholar] [CrossRef]
- Moravcová, D.; Kahle, V.; Rehulková, H.; Chmelík, J.; Rehulka, P. Short monolithic columns for purification and fractionation of peptide samples for matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometry analysis in proteomics. J. Chromatogr. A 2009, 1216, 3629–3636. [Google Scholar] [CrossRef]
- Franc, V.; Řehulka, P.; Medda, R.; Padiglia, A.; Floris, G.; Šebela, M. Analysis of the glycosylation pattern of plant copper amine oxidases by MALDI-TOF/TOF MS coupled to a manual chromatographic separation of glycans and glycopeptides. Electrophoresis 2013, 34, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Franc, V.; Řehulka, P.; Raus, M.; Stulík, J.; Novak, J.; Renfrow, M.B.; Šebela, M. Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: role of cysteine alkylation during sample processing. J. Proteom. 2013, 92, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franc, V.; Sebela, M.; Rehulka, P.; Končitíková, R.; Lenobel, R.; Madzak, C.; Kopečný, D. Analysis of N-glycosylation in maize cytokinin oxidase/dehydrogenase 1 using a manual microgradient chromatographic separation coupled offline to MALDI-TOF/TOF mass spectrometry. J. Proteom. 2012, 75, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- Rehulka, P.; Zahradnikova, M.; Rehulkova, H.; Dvorakova, P.; Nenutil, R.; Valik, D.; Vojtesek, B.; Hernychova, L.; Novotny, M.V. Microgradient separation technique for purification and fractionation of permethylated N-glycans before mass spectrometric analyses. J. Sep. Sci. 2018, 41, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Lenobel, R.; Rehulkova, H.; Sebela, M.; Franc, V.; Kahle, V.; Moravcova, D.; Rehulka, P. Analysis of peptide mixtures for proteomics research using LC–ESI-MS with a simple microgradient device. LC GC N. Am. 2015, 33, 420–428. [Google Scholar]
- Yang, F.; Shen, Y.; Camp, D.G.; Smith, R.D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteom. 2012, 9, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Batth, T.S.; Francavilla, C.; Olsen, J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. [Google Scholar] [CrossRef]
- Matsuda, H.; Nakamura, H.; Nakajima, T. New ceramic titania: selective adsorbent for organic phosphates. Anal. Sci. 1990, 6, 911–912. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chang-Wong, T.; Tang, H.-Y.; Speicher, D.W. Comparison of extensive protein fractionation and repetitive LC-MS/MS analyses on depth of analysis for complex proteomes. J. Proteome Res. 2010, 9, 1032–1040. [Google Scholar] [CrossRef]
- Yeh, T.-T.; Ho, M.-Y.; Chen, W.-Y.; Hsu, Y.-C.; Ku, W.-C.; Tseng, H.-W.; Chen, S.-T.; Chen, S.-F. Comparison of different fractionation strategies for in-depth phosphoproteomics by liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 3417–3424. [Google Scholar] [CrossRef]
- McNulty, D.E.; Annan, R.S. Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteom. 2008, 7, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, K.-Y.; Liu, H.-C.; Goshe, M.B. Development and application of a phosphoproteomic method using electrostatic repulsion-hydrophilic interaction chromatography (ERLIC), IMAC, and LC-MS/MS analysis to study Marek’s Disease Virus infection. J. Proteome Res. 2011, 10, 4041–4053. [Google Scholar] [CrossRef] [PubMed]
- Ritorto, M.S.; Cook, K.; Tyagi, K.; Pedrioli, P.G.A.; Trost, M. Hydrophilic strong anion exchange (hSAX) chromatography for highly orthogonal peptide separation of complex proteomes. J. Proteome Res. 2013, 12, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Batth, T.S.; Olsen, J.V. Offline high pH reversed-phase peptide fractionation for deep phosphoproteome coverage. Methods Mol. Biol. 2016, 1355, 179–192. [Google Scholar] [CrossRef]
- Snyder, L.R.; Kirkland, J.J.; Dolan, J.W. Ionic samples: Reversed-phase, ion-pair, and ion-exchange chromatography. In Introduction to Modern Liquid Chromatography; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; pp. 303–360. ISBN 978-0-470-50818-3. [Google Scholar]
- Kemp, B.E. Relative alkali stability of some peptide o-phosphoserine and o-phosphothreonine esters. FEBS Lett. 1980, 110, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Sickmann, A.; Meyer, H.E. Phosphoamino acid analysis. Proteomics 2001, 1, 200–206. [Google Scholar] [CrossRef]
- Kahle, V.; Vázlerová, M.; Welsch, T. Automated microgradient system for capillary electrochromatography. J. Chromatogr. A 2003, 990, 3–9. [Google Scholar] [CrossRef]
- Potel, C.M.; Lin, M.-H.; Heck, A.J.R.; Lemeer, S. Defeating major contaminants in Fe3+- immobilized metal ion affinity chromatography (IMAC) phosphopeptide enrichment. Mol. Cell. Proteom. 2018, 17, 1028–1034. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.-G.; Stanley, E.R. Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr. Protoc. Protein Sci. 2010, 59. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.R.; Thingholm, T.E.; Jensen, O.N.; Roepstorff, P.; Jørgensen, T.J.D. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteom. 2005, 4, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2009; Available online: https://www.R-project.org/ (accessed on 25 May 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | ||
|---|---|---|---|---|---|---|---|
| Conventional elution mobile phase | ACN | 400 | 320 | 240 | 160 | 80 | 20 |
| 200 mM HCOONH4 | 100 | 100 | 100 | 100 | 100 | 100 | |
| H2O | 500 | 580 | 660 | 740 | 820 | 880 | |
| ACN concentration | 40% | 32% | 24% | 16% | 8% | 2% | |
| Measured pH | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | |
| Optimized elution mobile phase | 1% acetic acid | 100 | 100 | 100 | 100 | 100 | 100 |
| 80% ACN/ 200 mM NH4OH | 300 | 250 | 200 | 150 | 100 | 50 | |
| H2O | 600 | 650 | 700 | 750 | 800 | 850 | |
| ACN concentration | 24% | 20% | 16% | 12% | 8% | 4% | |
| Measured pH | 6.57 | 5.64 | 5.19 | 4.80 | 4.42 | 3.92 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ondrej, M.; Rehulka, P.; Rehulkova, H.; Kupcik, R.; Tichy, A. Fractionation of Enriched Phosphopeptides Using pH/Acetonitrile-Gradient-Reversed-Phase Microcolumn Separation in Combination with LC–MS/MS Analysis. Int. J. Mol. Sci. 2020, 21, 3971. https://doi.org/10.3390/ijms21113971
Ondrej M, Rehulka P, Rehulkova H, Kupcik R, Tichy A. Fractionation of Enriched Phosphopeptides Using pH/Acetonitrile-Gradient-Reversed-Phase Microcolumn Separation in Combination with LC–MS/MS Analysis. International Journal of Molecular Sciences. 2020; 21(11):3971. https://doi.org/10.3390/ijms21113971
Chicago/Turabian StyleOndrej, Martin, Pavel Rehulka, Helena Rehulkova, Rudolf Kupcik, and Ales Tichy. 2020. "Fractionation of Enriched Phosphopeptides Using pH/Acetonitrile-Gradient-Reversed-Phase Microcolumn Separation in Combination with LC–MS/MS Analysis" International Journal of Molecular Sciences 21, no. 11: 3971. https://doi.org/10.3390/ijms21113971
APA StyleOndrej, M., Rehulka, P., Rehulkova, H., Kupcik, R., & Tichy, A. (2020). Fractionation of Enriched Phosphopeptides Using pH/Acetonitrile-Gradient-Reversed-Phase Microcolumn Separation in Combination with LC–MS/MS Analysis. International Journal of Molecular Sciences, 21(11), 3971. https://doi.org/10.3390/ijms21113971