Structure, Function, and Regulation of the SRMS Tyrosine Kinase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure and Activity of SRMS
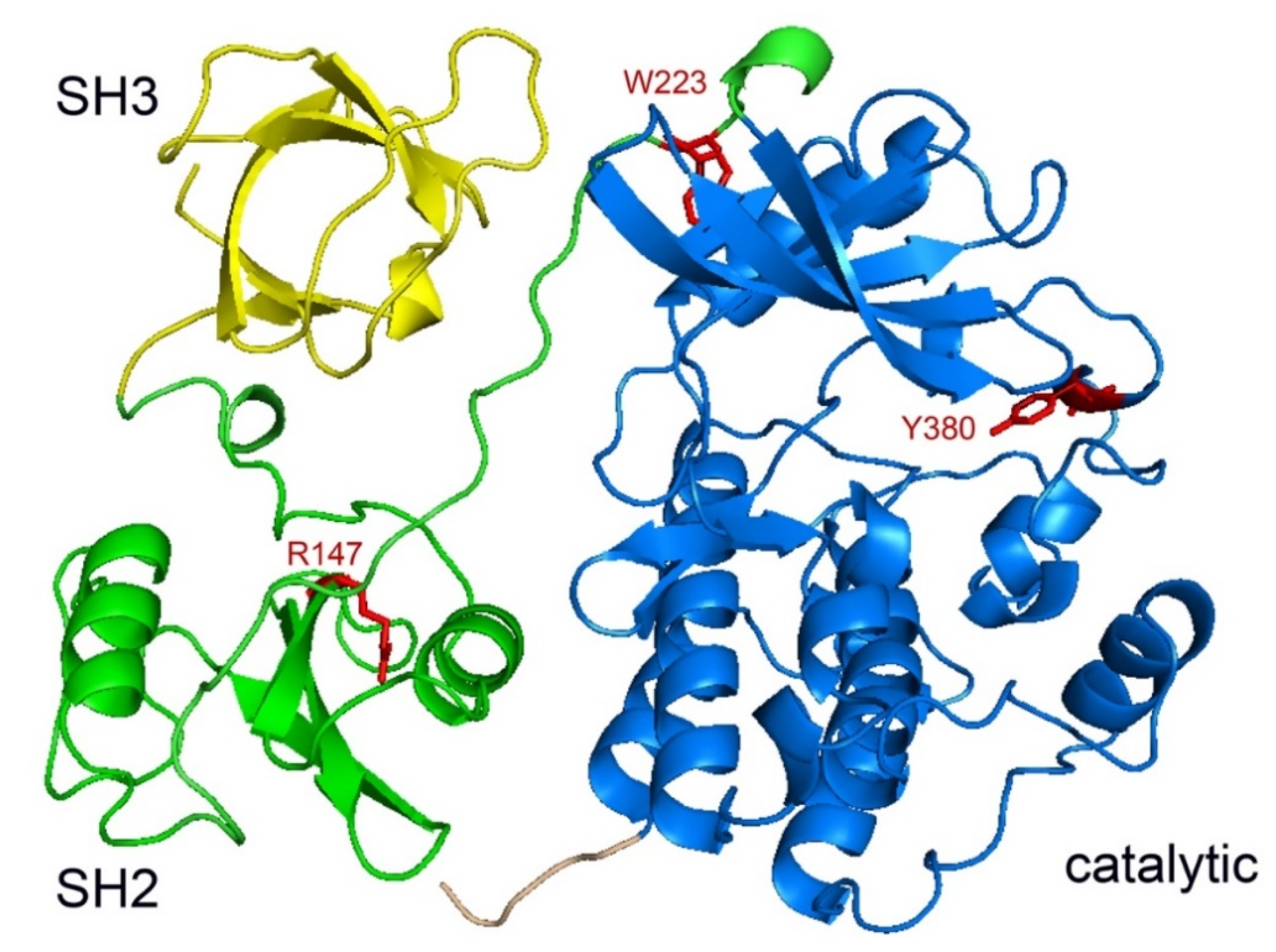
2.1. Domain Architecture
2.2. Structure of SRMS Kinase
2.3. In Vitro Activity of SRMS
3. Substrates of SRMS
4. Subcellular Localization of SRMS
5. Function of SRMS in Normal Cells
6. Function of SRMS in Human Disease
7. Summary
Funding
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| Brk | Breast tumor kinase |
| Dok1 | Docking protein 1 |
| EGF | Epidermal growth factor |
| Frk | Fyn-related kinase |
| GFP | Green fluorescent protein |
| HEK | Human embryonic kidney |
| LC-MS | Liquid chromatography-mass spectrometry |
| PTK6 | Protein tyrosine kinase 6 |
| SFK | Src family kinase |
| SH2 | Src homology 2 |
| SH3 | Src homology 3 |
| siRNA | Small interfering RNA |
| SRMS | Src-related kinase lacking C-terminal regulatory tyrosine and N-terminal myristoylation sites |
References
- Kohmura, N.; Yagi, T.; Tomooka, Y.; Oyanagi, M.; Kominami, R.; Takeda, N.; Chiba, J.; Ikawa, Y.; Aizawa, S. A novel nonreceptor tyrosine kinase, Srm: Cloning and targeted disruption. Mol. Cell Biol. 1994, 14, 6915–6925. [Google Scholar] [CrossRef] [PubMed]
- Kawachi, Y.; Nakauchi, H.; Otsuka, F. Isolation of a cDNA encoding a tyrosine kinase expressed in murine skin. Exp. Dermatol. 1997, 6, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Serfas, M.S.; Tyner, A.L. Brk, Srm, Frk, and Src42A form a distinct family of intracellular Src-like tyrosine kinases. Oncol. Res. 2003, 13, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Brauer, P.M.; Tyner, A.L. Building a better understanding of the intracellular tyrosine kinase PTK6—BRK by BRK. Biochim. Biophys. Acta 2010, 1806, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrander, J.H.; Daniel, A.R.; Lange, C.A. Brk/PTK6 signaling in normal and cancer cell models. Curr. Opin. Pharmacol. 2010, 10, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Lee, K.H.; Kim, H.; Lee, S.T. Assignment of the human PTK6 gene encoding a non-receptor protein tyrosine kinase to 20q13.3 by fluorescence in situ hybridization. Cytogenet. Cell Genet. 1997, 77, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Aleem, S.; Yang, M.; Miller, W.T.; Tonks, N.K. Protein-tyrosine phosphatase and kinase specificity in regulation of SRC and breast tumor kinase. J. Biol. Chem. 2015, 290, 15934–15947. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, J.G.; Chin, K.; Collins, C.; Gray, J.W. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res. Treat. 2003, 78, 337–345. [Google Scholar] [CrossRef]
- Goel, R.K.; Miah, S.; Black, K.; Kalra, N.; Dai, C.; Lukong, K.E. The unique N-terminal region of SRMS regulates enzymatic activity and phosphorylation of its novel substrate docking protein 1. FEBS J. 2013, 280, 4539–4559. [Google Scholar] [CrossRef]
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1996, 1287, 121–149. [Google Scholar] [CrossRef]
- Mayer, B.J. Signal transduction: Clamping down on Src activity. Curr. Biol. 1997, 7, R295–R298. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.A.; Howell, B. The when and how of Src regulation. Cell 1993, 73, 1051–1054. [Google Scholar] [CrossRef]
- Yadav, S.S.; Miller, W.T. The evolutionarily conserved arrangement of domains in SRC family kinases is important for substrate recognition. Biochemistry 2008, 47, 10871–10880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.H.; Amacher, J.F.; Nocka, L.M.; Kuriyan, J. The Src module: An ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 535–563. [Google Scholar] [CrossRef] [Green Version]
- Suga, H.; Miller, W.T. Src signaling in a low-complexity unicellular kinome. Sci. Rep. 2018, 8, 5362. [Google Scholar] [CrossRef] [Green Version]
- Suga, H.; Torruella, G.; Burger, G.; Brown, M.W.; Ruiz-Trillo, I. Earliest Holozoan expansion of phosphotyrosine signaling. Mol. Biol. Evol. 2014, 31, 517–528. [Google Scholar] [CrossRef] [Green Version]
- D’Aniello, S.; Irimia, M.; Maeso, I.; Pascual-Anaya, J.; Jimenez-Delgado, S.; Bertrand, S.; Garcia-Fernandez, J. Gene expansion and retention leads to a diverse tyrosine kinase superfamily in amphioxus. Mol. Biol. Evol. 2008, 25, 1841–1854. [Google Scholar] [CrossRef] [Green Version]
- Buss, J.E.; Kamps, M.P.; Gould, K.; Sefton, B.M. The absence of myristic acid decreases membrane binding of p60src but does not affect tyrosine protein kinase activity. J. Virol. 1986, 58, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Kamps, M.P.; Buss, J.E.; Sefton, B.M. Rous sarcoma virus transforming protein lacking myristic acid phosphorylates known polypeptide substrates without inducing transformation. Cell 1986, 45, 105–112. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharm. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Bromann, P.A.; Korkaya, H.; Courtneidge, S.A. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 2004, 23, 7957–7968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Alsaidan, O.A.; Goodwin, O.; Li, Q.; Sulejmani, E.; Han, Z.; Bai, A.; Albers, T.; Beharry, Z.; Zheng, Y.G.; et al. Blocking myristoylation of Src inhibits its kinase activity and suppresses prostate cancer progression. Cancer Res. 2017, 77, 6950–6962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [Green Version]
- Arbesu, M.; Maffei, M.; Cordeiro, T.N.; Teixeira, J.M.; Perez, Y.; Bernado, P.; Roche, S.; Pons, M. The unique domain forms a fuzzy intramolecular complex in Src family kinases. Structure 2017, 25, 630–640.e634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, E.T.; Kezdy, F.J. Secondary structures of proteins and peptides in amphiphilic environments. (A review). Proc. Natl. Acad. Sci. USA 1983, 80, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Arbesu, M.; Pons, M. Integrating disorder in globular multidomain proteins: Fuzzy sensors and the role of SH3 domains. Arch. Biochem. Biophys. 2019, 677, 108161. [Google Scholar] [CrossRef]
- Kuriyan, J.; Cowburn, D. Modular peptide recognition domains in eukaryotic signaling. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 259–288. [Google Scholar] [CrossRef]
- Mayer, B.J. The discovery of modular binding domains: Building blocks of cell signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 691–698. [Google Scholar] [CrossRef]
- Miller, W.T. Determinants of substrate recognition in nonreceptor tyrosine kinases. Acc. Chem. Res. 2003, 36, 393–400. [Google Scholar] [CrossRef]
- Sicheri, F.; Kuriyan, J. Structures of Src-family tyrosine kinases. Curr. Opin. Struct. Biol. 1997, 7, 777–785. [Google Scholar] [CrossRef]
- Briggs, S.D.; Sharkey, M.; Stevenson, M.; Smithgall, T.E. SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 1997, 272, 17899–17902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moarefi, I.; LaFevre-Bernt, M.; Sicheri, F.; Huse, M.; Lee, C.H.; Kuriyan, J.; Miller, W.T. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 1997, 385, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.J.; Baltimore, D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol. Cell Biol. 1994, 14, 2883–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, H.; Miller, W.T. Regulation of the nonreceptor tyrosine kinase brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 2002, 277, 34634–34641. [Google Scholar] [CrossRef] [Green Version]
- Goel, R.K.; Paczkowska, M.; Reimand, J.; Napper, S.; Lukong, K.E. phosphoproteomics analysis identifies novel candidate substrates of the nonreceptor tyrosine kinase, Src-related kinase lacking C-terminal regulatory tyrosine and N-terminal myristoylation sites (SRMS). Mol. Cell Proteom. 2018, 17, 925–947. [Google Scholar] [CrossRef] [Green Version]
- Hennequin, L.F.; Allen, J.; Breed, J.; Curwen, J.; Fennell, M.; Green, T.P.; Lambert-van der Brempt, C.; Morgentin, R.; Norman, R.A.; Olivier, A.; et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem. 2006, 49, 6465–6488. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2016, 86. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src [see comments]. Nature 1997, 385, 595–602. [Google Scholar] [CrossRef]
- LaFevre-Bernt, M.; Sicheri, F.; Pico, A.; Porter, M.; Kuriyan, J.; Miller, W.T. Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. J. Biol. Chem. 1998, 273, 32129–32134. [Google Scholar] [CrossRef] [Green Version]
- Okada, M. Regulation of the SRC family kinases by Csk. Int. J. Biol. Sci. 2012, 8, 1385–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenglowsky, S.; Ahrendt, K.A.; Buckmelter, A.J.; Feng, B.; Gloor, S.L.; Gradl, S.; Grina, J.; Hansen, J.D.; Laird, E.R.; Lunghofer, P.; et al. Pyrazolopyridine inhibitors of B-RafV600E. Part 2: Structure-activity relationships. Bioorg. Med. Chem. Lett. 2011, 21, 5533–5537. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, J. New roles for Src kinases in control of cell survival and angiogenesis. Cell 2000, 100, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Goel, R.K.; Meyer, M.; Paczkowska, M.; Reimand, J.; Vizeacoumar, F.; Vizeacoumar, F.; Lam, T.T.; Lukong, K.E. Global phosphoproteomic analysis identifies SRMS-regulated secondary signaling intermediates. Proteome Sci. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Li, B.; Sara, A.; Ay, C.; Leung, W.Y.; Zhang, Y.; Dong, Y.; Liang, Q.; Zhang, X.; Weidner, P.; et al. Docking protein-1 promotes inflammatory macrophage signaling in gastric cancer. Oncoimmunology 2019, 8, e1649961. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Kawamura, Y.; Miura, A.; Mori, M.; Wakamatsu, A.; Yamamoto, J.; Isogai, T.; Matsumoto, M.; Nakayama, K.I.; Natsume, T.; et al. Comparative analysis of human SRC-family kinase substrate specificity in vitro. J. Proteome Res. 2010, 9, 5982–5993. [Google Scholar] [CrossRef]
- Noguchi, T.; Matozaki, T.; Inagaki, K.; Tsuda, M.; Fukunaga, K.; Kitamura, Y.; Kitamura, T.; Shii, K.; Yamanashi, Y.; Kasuga, M. Tyrosine phosphorylation of p62(Dok) induced by cell adhesion and insulin: Possible role in cell migration. EMBO J. 1999, 18, 1748–1760. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Wisniewski, D.; Strife, A.; Shivakrupa; Clarkson, B.; Resh, M.D. Phosphatidylinositol 3-kinase and Src family kinases are required for phosphorylation and membrane recruitment of Dok-1 in c-Kit signaling. J. Biol. Chem. 2002, 277, 13732–13738. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Alicea-Velazquez, N.L.; Bannwarth, L.; Lehtonen, S.I.; Boggon, T.J.; Cheng, H.C.; Hytonen, V.P.; Turk, B.E. Global analysis of human nonreceptor tyrosine kinase specificity using high-density peptide microarrays. J. Proteome Res. 2014, 13, 4339–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivaska, J.; Pallari, H.M.; Nevo, J.; Eriksson, J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Resnick, R.J.; Shalloway, D. Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC Cell Biol. 2004, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derry, J.J.; Richard, S.; Carvajal, H.V.; Ye, X.; Vasioukhin, V.; Cochrane, A.W.; Chen, T.; Tyner, A.L. Sik (BRK) phosphorylates Sam68 in the nucleus and negatively regulates its RNA binding ability. Mol. Cell Biol. 2000, 20, 6114–6126. [Google Scholar] [CrossRef] [Green Version]
- Derry, J.J.; Prins, G.S.; Ray, V.; Tyner, A.L. Altered localization and activity of the intracellular tyrosine kinase BRK/Sik in prostate tumor cells. Oncogene 2003, 22, 4212–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roux, A.L.; Busquets, M.A.; Sagues, F.; Pons, M. Kinetics characterization of c-Src binding to lipid membranes: Switching from labile to persistent binding. Colloids Surf. B Biointerfaces 2016, 138, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.M.; Rentero, C.; Rossy, J.; Magenau, A.; Williamson, D.; Rodriguez, M.; Gaus, K. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. J. Biophotonics 2010, 3, 446–454. [Google Scholar] [CrossRef]
- Smith, A.W.; Huang, H.H.; Endres, N.F.; Rhodes, C.; Groves, J.T. Dynamic organization of myristoylated Src in the live cell plasma membrane. J. Phys. Chem. B 2016, 120, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Potts, M.B.; Kim, H.S.; Fisher, K.W.; Hu, Y.; Carrasco, Y.P.; Bulut, G.B.; Ou, Y.H.; Herrera-Herrera, M.L.; Cubillos, F.; Mendiratta, S.; et al. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci. Signal. 2013, 6, ra90. [Google Scholar] [CrossRef] [Green Version]
- Buffard, M.; Naldi, A.; Radulescu, O.; Coopman, P.J.; Larive, R.M.; Freiss, G. Network reconstruction and significant pathway extraction using phosphoproteomic data from cancer cells. Proteomics 2019, 19, e1800450. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.J.; Barker, K.T.; Martindale, J.E.; Kamalati, T.; Lowe, P.N.; Page, M.J.; Gusterson, B.A.; Crompton, M.R. Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 1994, 9, 2383–2390. [Google Scholar] [PubMed]
- Barker, K.T.; Jackson, L.E.; Crompton, M.R. BRK tyrosine kinase expression in a high proportion of human breast carcinomas. Oncogene 1997, 15, 799–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, B.; Chatti, K.; Qiu, H.; Lakshmi, B.; Krasnitz, A.; Hicks, J.; Yu, M.; Miller, W.T.; Muthuswamy, S.K. Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12463–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, M.W.; Park, J.; Han, H.S.; Yun, Y.M.; Kang, J.W.; Choi, D.Y.; Lee, J.W.; Jung, J.H.; Lee, K.Y.; Kim, K.P. Discovery of gastric cancer specific biomarkers by the application of serum proteomics. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Eschrich, S.; Li, J.; Bai, Y.; Koomen, J.; Mayer, B.J.; Haura, E.B. Characterizing tyrosine phosphorylation signaling in lung cancer using SH2 profiling. PLoS ONE 2010, 5, e13470. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.P.; Willey, C.D.; Anderson, J.C.; Welaya, K.; Chen, D.; Mehta, A.; Ghatalia, P.; Madan, A.; Naik, G.; Sudarshan, S.; et al. Kinomic profiling identifies focal adhesion kinase 1 as a therapeutic target in advanced clear cell renal cell carcinoma. Oncotarget 2017, 8, 29220–29232. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Creixell, P.; Schoof, E.M.; Simpson, C.D.; Longden, J.; Miller, C.J.; Lou, H.J.; Perryman, L.; Cox, T.R.; Zivanovic, N.; Palmeri, A.; et al. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 2015, 163, 202–217. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McClendon, C.J.; Miller, W.T. Structure, Function, and Regulation of the SRMS Tyrosine Kinase. Int. J. Mol. Sci. 2020, 21, 4233. https://doi.org/10.3390/ijms21124233
McClendon CJ, Miller WT. Structure, Function, and Regulation of the SRMS Tyrosine Kinase. International Journal of Molecular Sciences. 2020; 21(12):4233. https://doi.org/10.3390/ijms21124233
Chicago/Turabian StyleMcClendon, Chakia J., and W. Todd Miller. 2020. "Structure, Function, and Regulation of the SRMS Tyrosine Kinase" International Journal of Molecular Sciences 21, no. 12: 4233. https://doi.org/10.3390/ijms21124233
APA StyleMcClendon, C. J., & Miller, W. T. (2020). Structure, Function, and Regulation of the SRMS Tyrosine Kinase. International Journal of Molecular Sciences, 21(12), 4233. https://doi.org/10.3390/ijms21124233