Characterization and Analysis of the Full-Length Transcriptomes of Multiple Organs in Pseudotaxus chienii (W.C.Cheng) W.C.Cheng

Abstract
:1. Introduction
2. Results
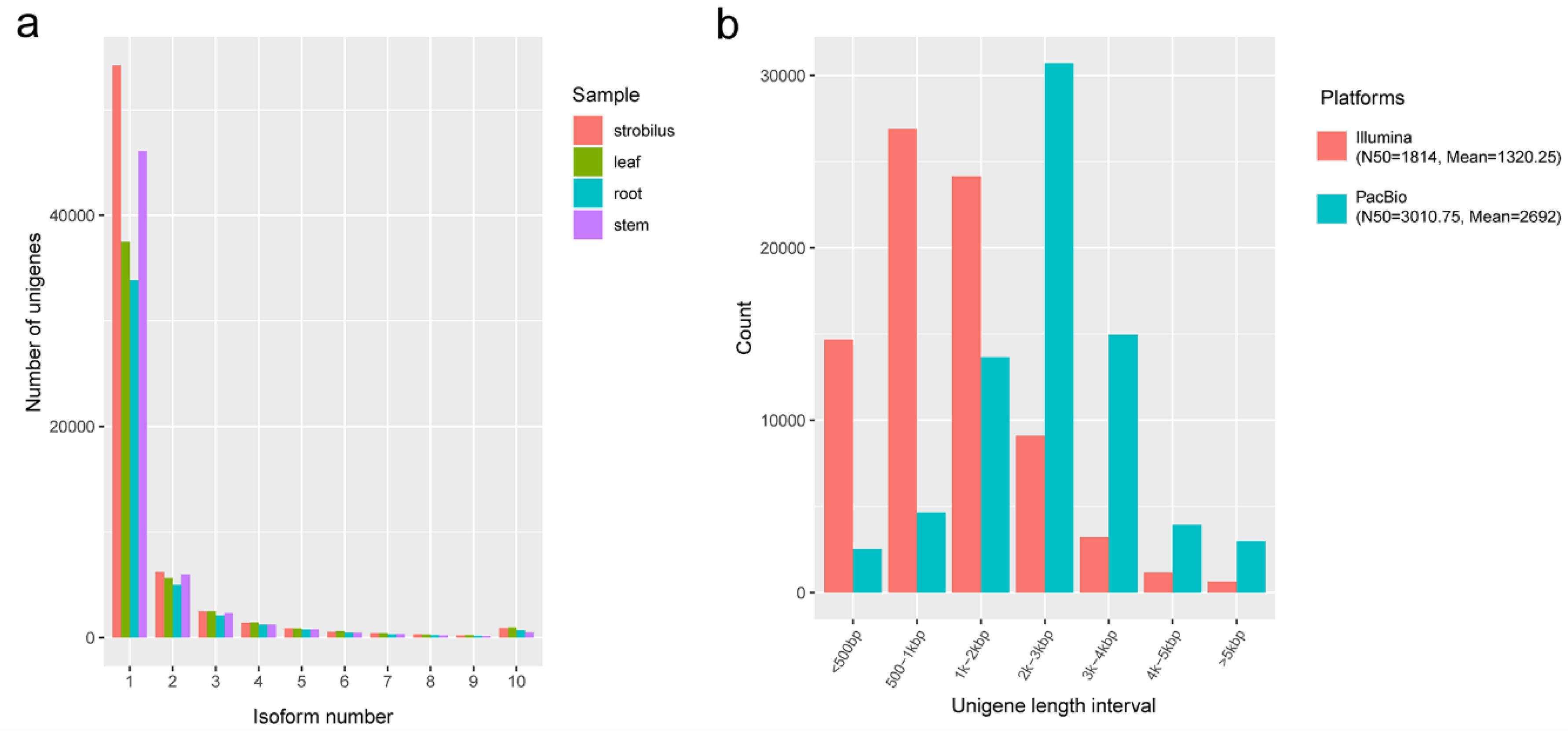
2.1. The Full-Length Sequences of PacBio Iso-Seq
2.2. De Novo Assembly of Illumina RNA-Seq Data
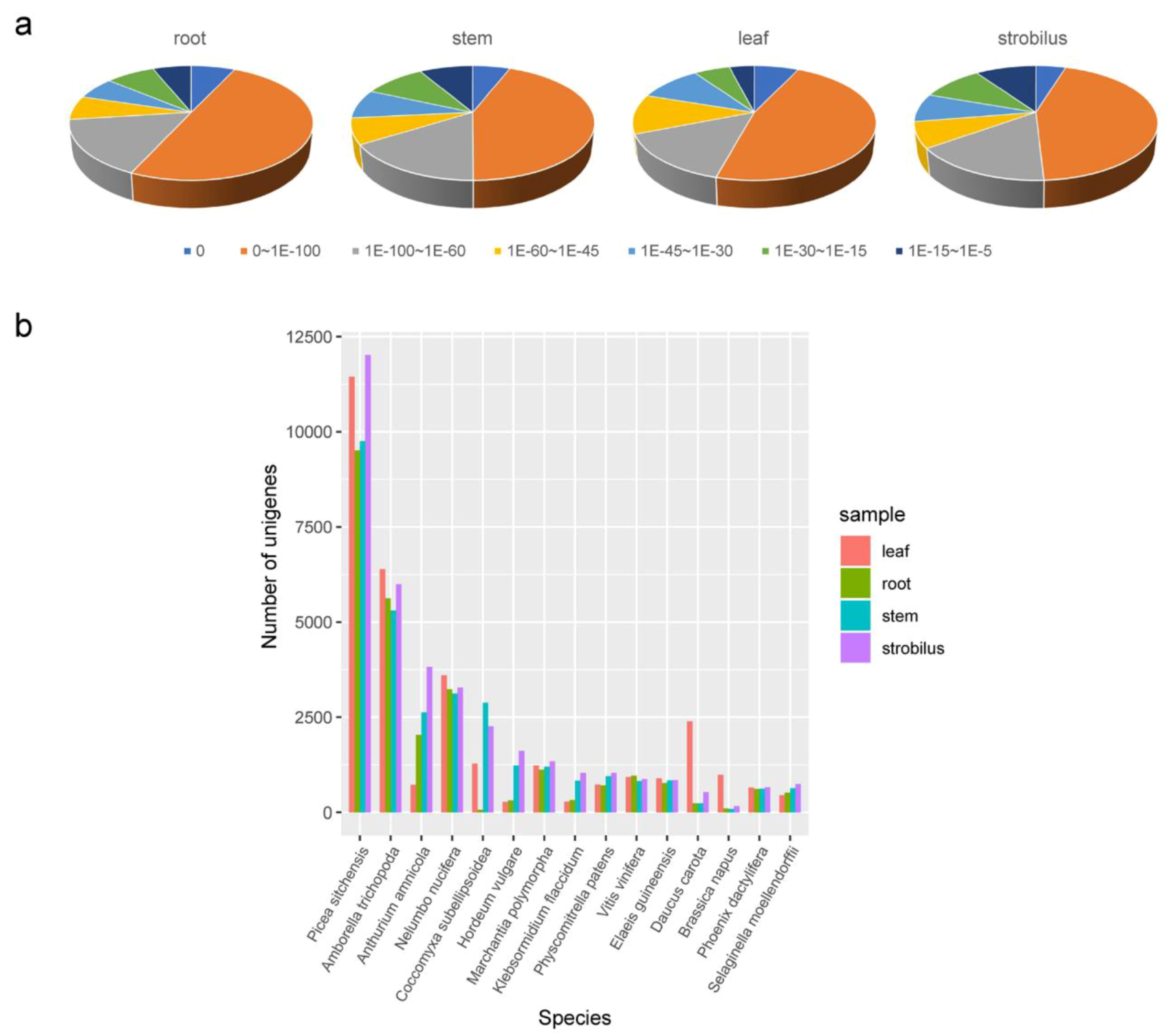
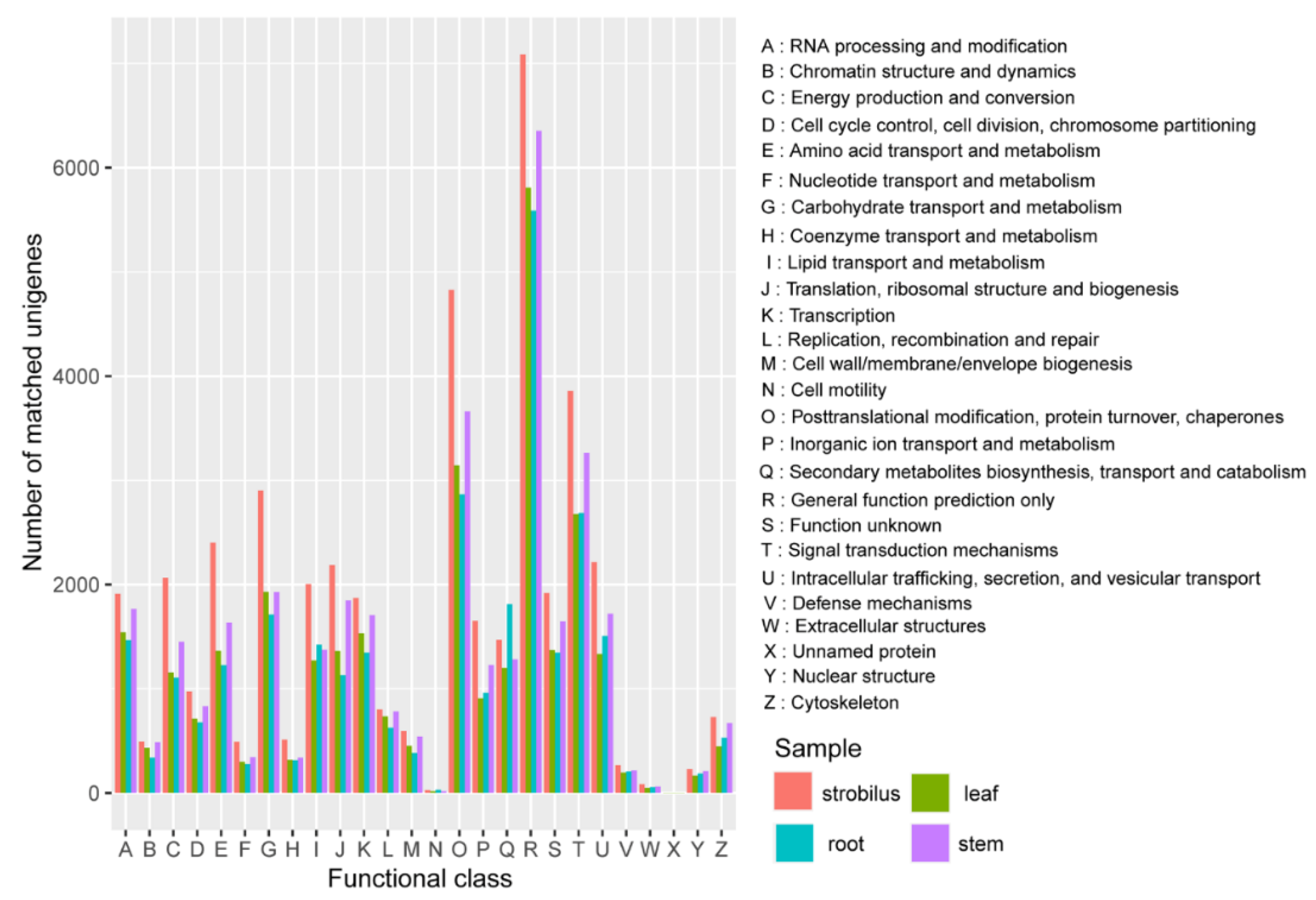
2.3. Functional Annotation
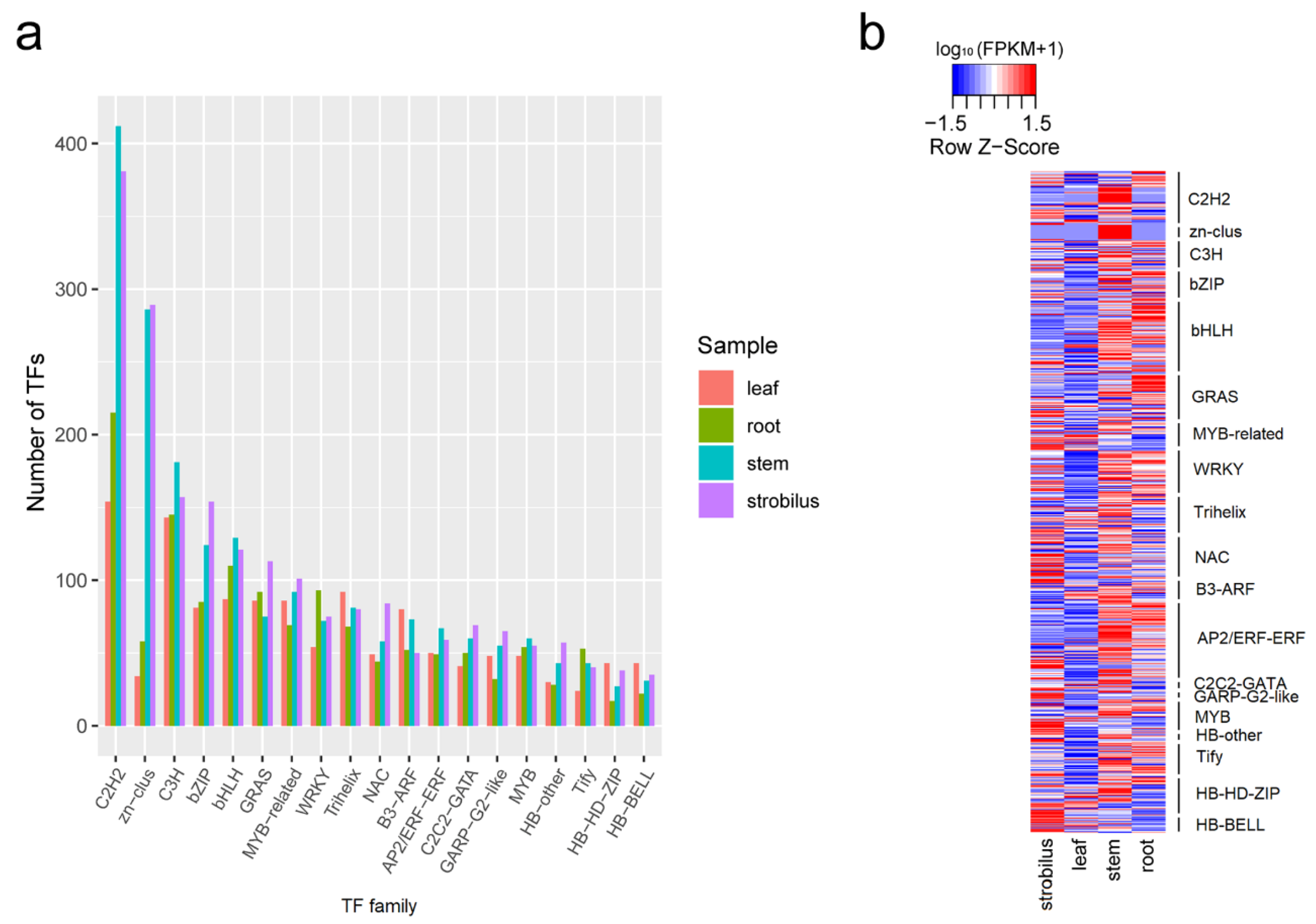
2.4. Identification of CDSs, TFs, and LncRNAs
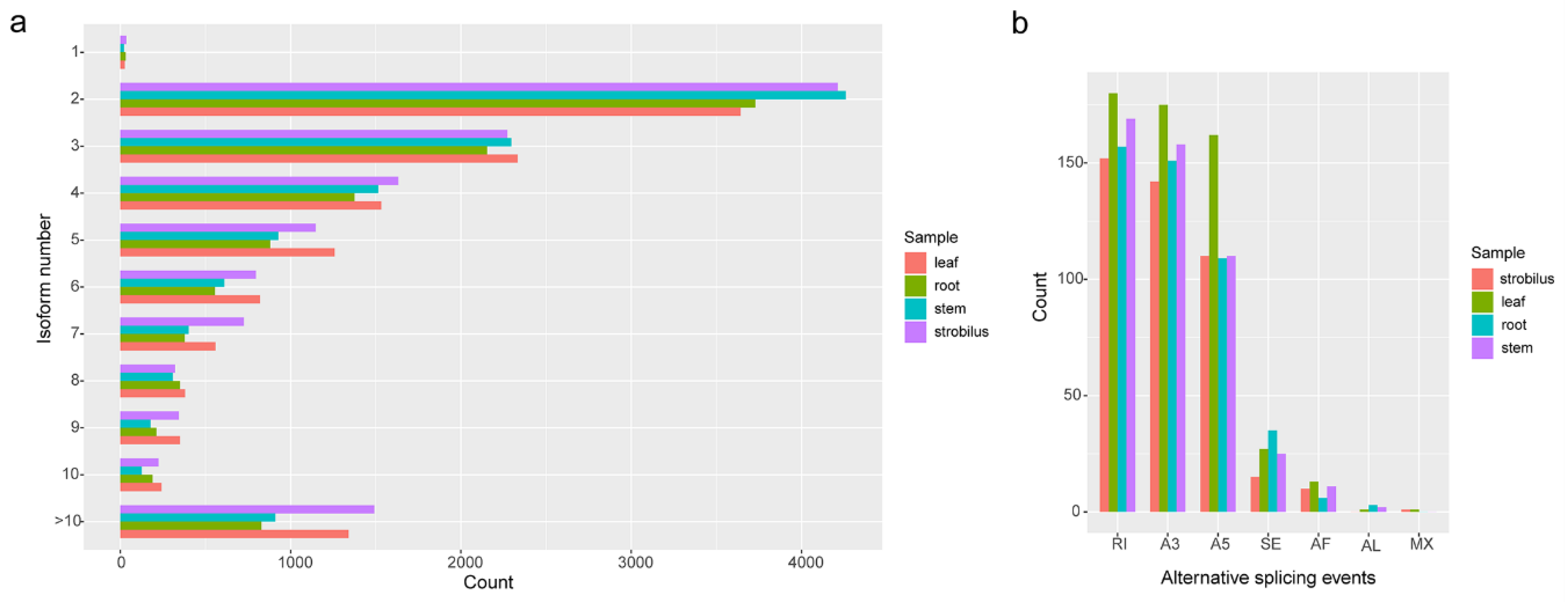
2.5. AS Analysis
2.6. Gene Expression Quantification
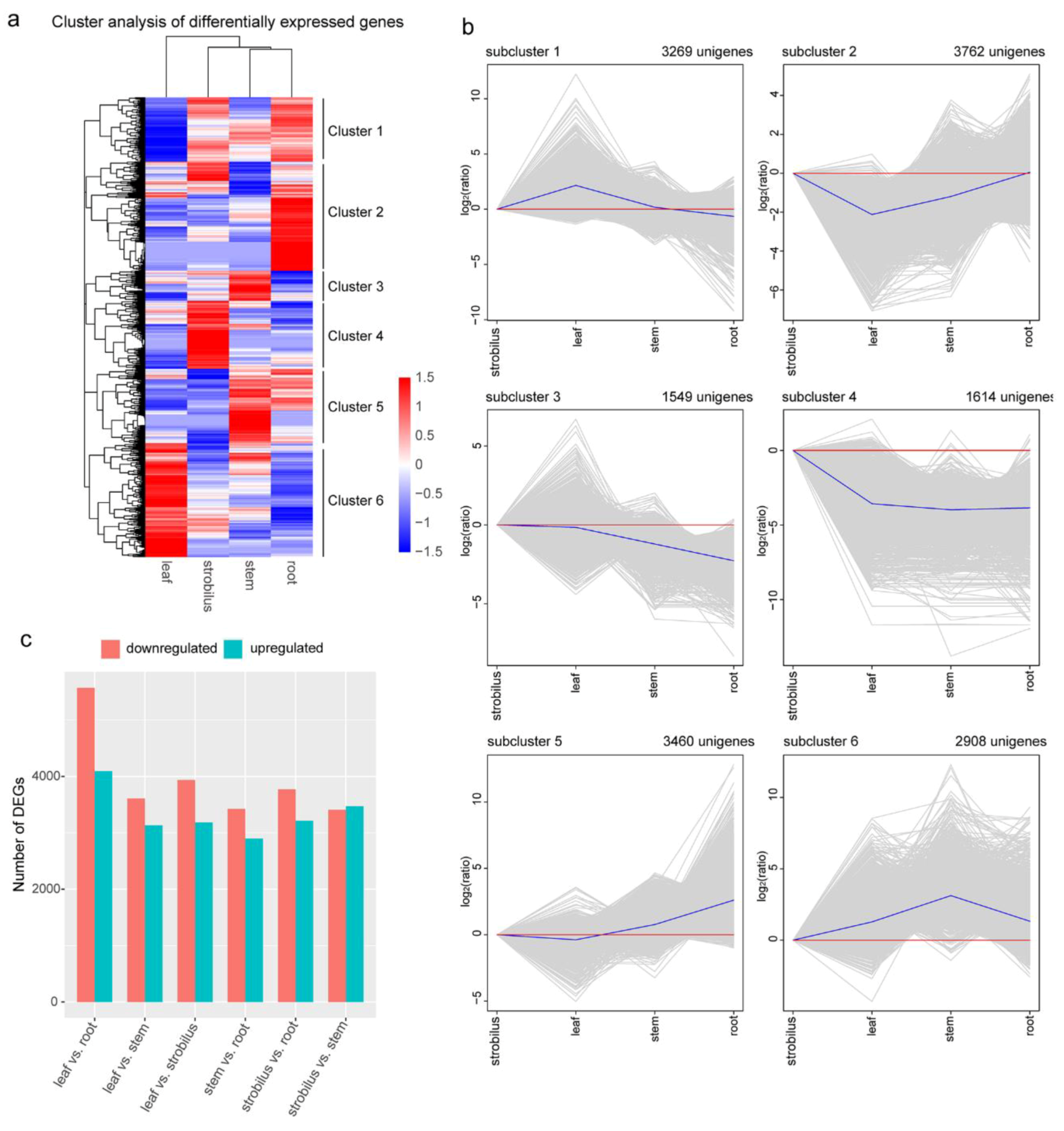
2.7. DEGs and Functional Enrichment Analysis
2.8. Organ-Specific Expression Unigenes
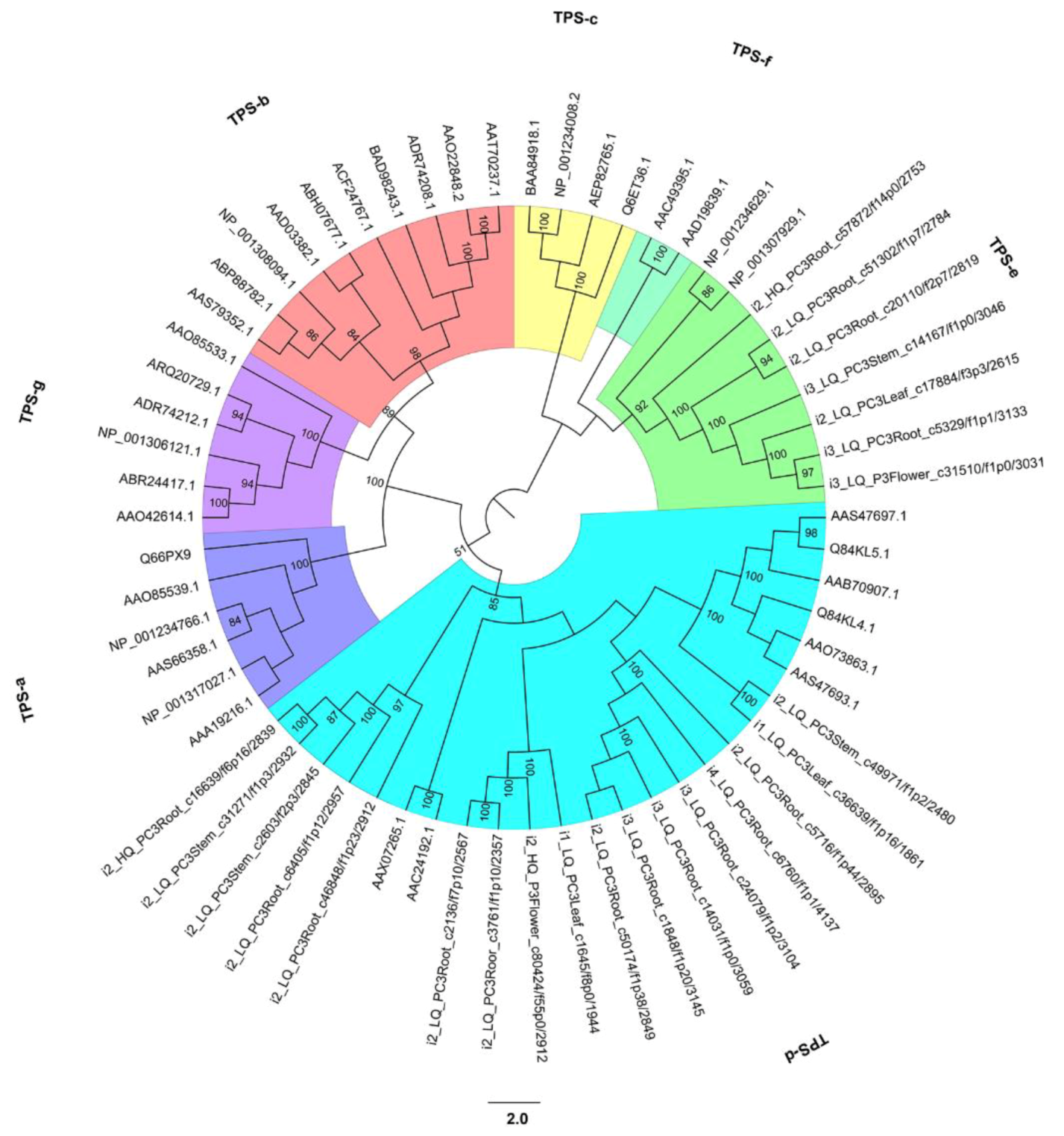
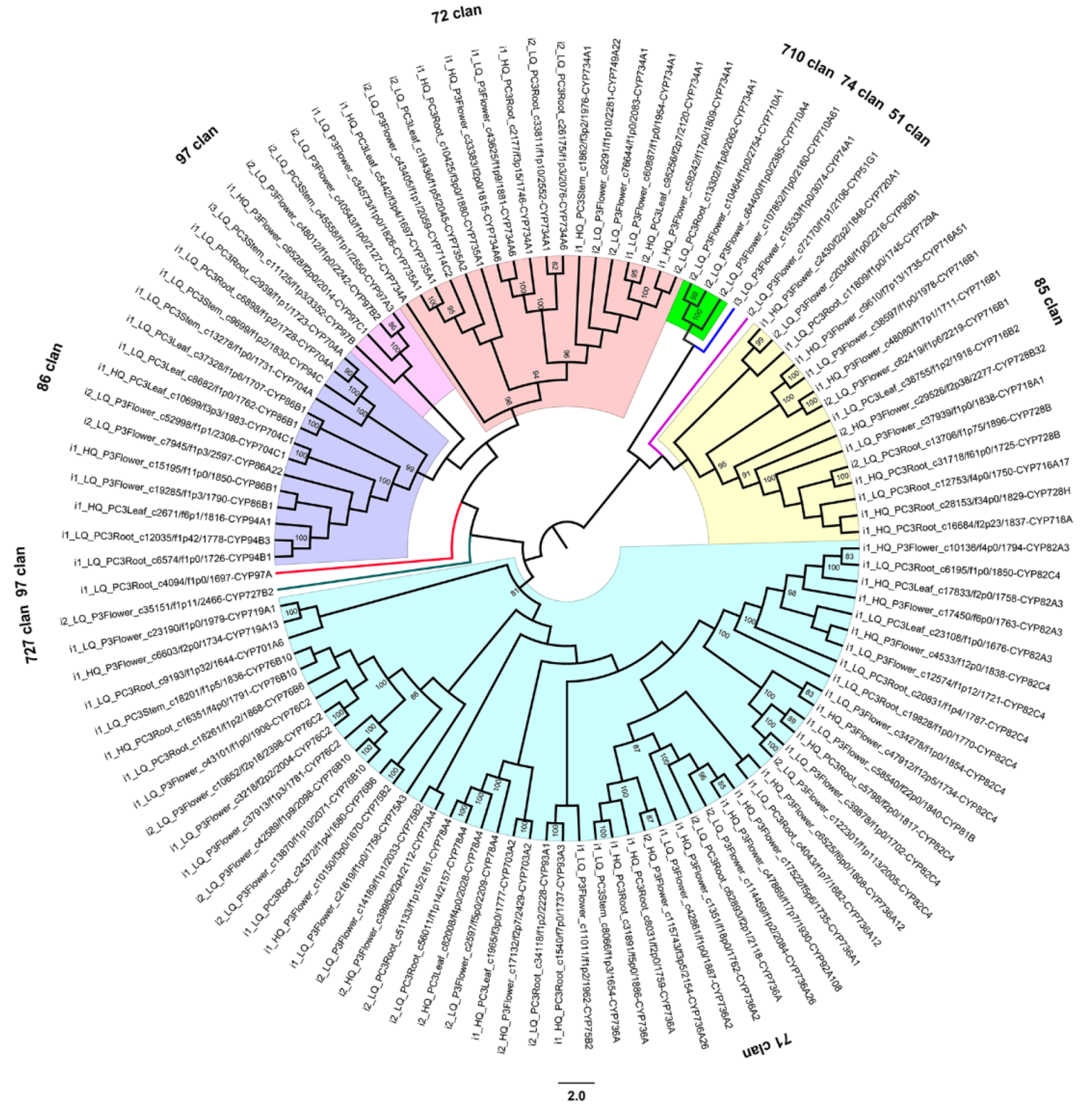
2.9. Discovery of Gene Families Related to Biotic/Abiotic Factors in P. chienii
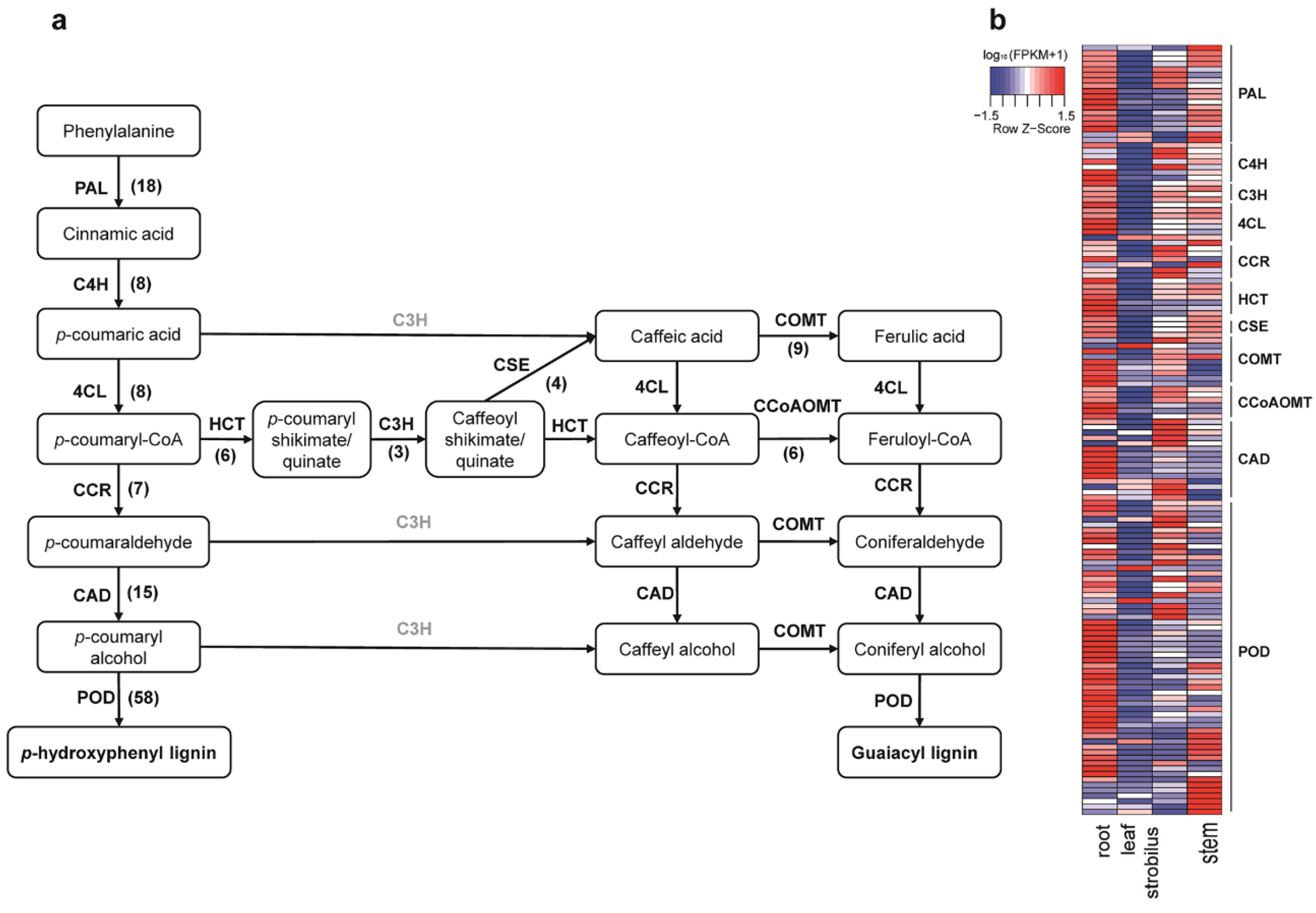
2.10. Characterization of the Unigenes in Phenylpropanoid Biosynthesis Pathway
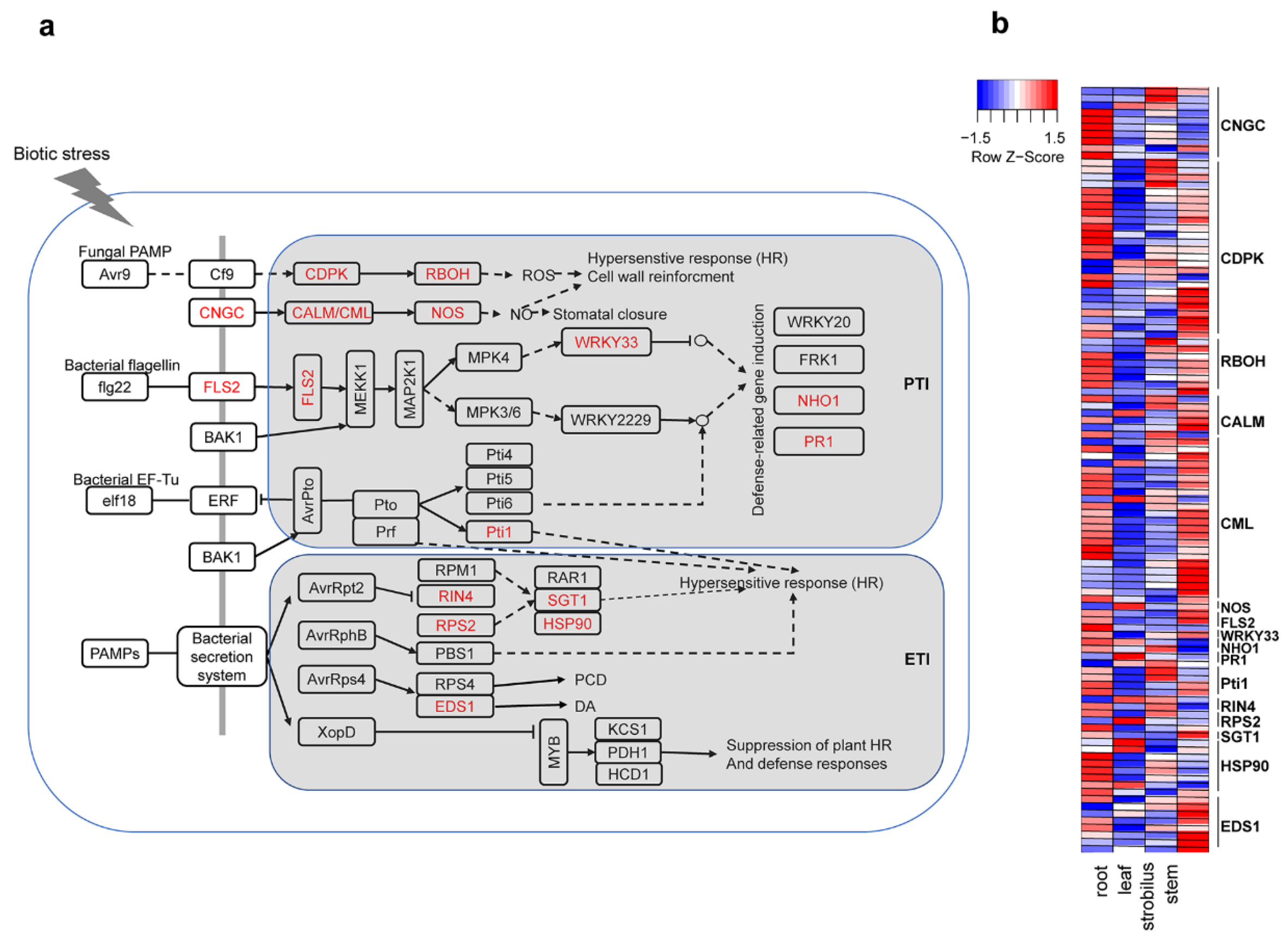
2.11. Characterization of the Unigenes in Plant–Pathogen Interactions
3. Discussion
3.1. PacBio Long Reads vs. Illumina Short Reads
3.2. Functional Annotation
3.3. TFs, LncRNAs, and AS Analysis
3.4. DEGs and Organ-Specific Unigenes
3.5. Gene Families Related to Biotic/Abiotic Factors in P. chienii
3.6. Characterization of the Unigenes in Phenylpropanoid Biosynthesis Pathway
3.7. Characterization of the Unigenes in Plant–Pathogen Interactions
4. Materials and Methods
4.1. Plant Materials and RNA Isolation
4.2. PacBio Library Construction and Sequencing
4.3. Illumina Library Construction, Sequencing, and De Novo Assembly
4.4. Preprocessing of the PacBio Iso-Seq Data
4.5. Functional Annotation
4.6. Prediction of CDSs, TFs, and LncRNAs
4.7. AS Analysis
4.8. Gene Expression Quantification, DEGs, and Functional Enrichment Analysis
4.9. Identification of the Gene Families Related to Biotic/Abiotic Factors
4.10. Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fu, L.G.; Li, N.; Mill, R.R. Taxaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999; Volume 4, pp. 89–98. [Google Scholar]
- Fu, L.G.; Jin, J.M. Red List of Endangered Plants in China; Science Press: Beijing, China, 1992. [Google Scholar]
- Lin, J.X.; He, X.Q.; Hu, Y.S. White berry yew (Pseudotaxus chienii (WC Cheng) WC Cheng). In Conifers: Status Survey and Conservation Action Plan; IUCN: Gland, Switzerland; Cambridge, UK, 1999; p. 106. [Google Scholar]
- Thomas, P.; Yang, Y. Pseudotaxus chienii. The IUCN Red List of Threatened Species 2013: e.T32798A2823334. Available online: https://dx.doi.org/10.2305/IUCN.UK.2013-1.RLTS.T32798A2823334.en (accessed on 16 June 2020).
- Su, Y.; Wang, T.; Ouyang, P. High genetic differentiation and variation as revealed by ISSR marker in Pseudotaxus chienii (Taxaceae), an old rare conifer endemic to China. Biochem. Syst. Ecol. 2009, 37, 579–588. [Google Scholar] [CrossRef]
- Hilfiker, K.; Gugerli, F.; Schutz, J.P.; Rotach, P.; Holderegger, R. Low RAPD variation and female-biased sex ratio indicate genetic drift in small populations of the dioecious conifer Taxus baccata in Switzerland. Conserv. Genet. 2004, 5, 357–365. [Google Scholar] [CrossRef]
- Wang, S. Functional Characterization and Evolutionary Analysis of Taxadiene Synthase Gene in Plants. Ph.D. Thesis, Chinese Academy of Forestry, Beijing, China, 2016. [Google Scholar]
- Ma, L.; Sun, N.; Liu, X.; Jiao, Y.; Zhao, H.; Deng, X.W. Organ-specific expression of Arabidopsis genome during development. Plant Physiol. 2005, 138, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raherison, E.S.; Giguère, I.; Caron, S.; Lamara, M.; MacKay, J.J. Modular organization of the white spruce (Picea glauca) transcriptome reveals functional organization and evolutionary signatures. New Phytol. 2015, 207, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Cañas, R.A.; Li, Z.; Pascual, M.B.; Castro-Rodríguez, V.; Ávila, C.; Sterck, L.; Van de Peer, Y.; Canovas, F.M. The gene expression landscape of pine seedling tissues. Plant J. 2017, 91, 1064–1087. [Google Scholar] [CrossRef] [Green Version]
- Yaqoob, N.; Yakovlev, I.A.; Krokene, P.; Kvaalen, H.; Solheim, H.; Fossdal, C.G. Defence-related gene expression in bark and sapwood of Norway spruce in response to Heterobasidion parviporum and methyl jasmonate. Physiol. Mol. Plant Pathol. 2012, 77, 10–16. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Kissling, R.; Gugerli, F.; Johnsen, O.; Skroppa, T.; Cheddadi, R.; van Der Knaap, W.O.; Latalowa, M.; Terhuerne-Berson, R.; Litt, T.; et al. Genetic consequences of glacial survival and postglacial colonization in Norway spruce: Combined analysis of mitochondrial DNA and fossil pollen. Mol. Ecol. 2008, 17, 4134–4150. [Google Scholar] [CrossRef]
- Phillips, M.A.; Croteau, R.B. Resin-based defenses in conifers. Trends Plant Sci. 1999, 4, 184–190. [Google Scholar] [CrossRef]
- Kelly, R.; Chipman, M.L.; Higuera, P.E.; Stefanova, I.; Brubaker, L.B.; Hu, F.S. Recent burning of boreal forests exceeds fire regime limits of the past 10,000 years. Proc. Natl. Acad. Sci. USA 2013, 110, 13055–13060. [Google Scholar] [CrossRef] [Green Version]
- Rejeb, I.B.; Pastor, V.; Mauch-Mani, B. Plant responses to simultaneous biotic and abiotic stress: Molecular mechanisms. Plants 2014, 3, 458–475. [Google Scholar] [CrossRef]
- Hall, D.E.; Yuen, M.M.S.; Jancsik, S.; Quesada, A.L.; Dullat, H.K.; Li, M.; Henderson, H.; Arango-Velez, A.; Liao, N.Y.; Docking, R.T.; et al. Transcriptome resources and functional characterization of monoterpene synthases for two host species of the mountain pine beetle, lodgepole pine (Pinus contorta) and jack pine (Pinus banksiana). BMC Plant Biol. 2013, 13, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlmann, J. Pine terpenoid defences in the mountain pine beetle epidemic and in other conifer pest interactions: Specialized enemies are eating holes into a diverse, dynamic and durable defence system. Tree Physiol. 2012, 32, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.A. Plant cytochrome P450 monooxygenases. Crit. Rev. Plant Sci. 1996, 15, 235–284. [Google Scholar] [CrossRef]
- Lorenz, W.W.; Sun, F.; Liang, C.; Kolychev, D.; Wang, H.M.; Zhao, X.; Cordonnier-Pratt, M.M.; Pratt, L.H.; Dean, J.F.D. Water stress-responsive genes in loblolly pine (Pinus taeda) roots identified by analyses of expressed sequence tag libraries. Tree Physiol. 2006, 26, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkinson, J.I.; Sioson, A.A.; Vasquez-Robinet, C.; Shukla, M.; Kumar, D.; Ellis, M.; Heath, L.S.; Ramakrishnan, N.; Chevone, B.; Watson, L.T.; et al. Photosynthetic acclimation is reflected in specific patterns of gene expression in drought-stressed loblolly pine. Plant Physiol. 2003, 133, 1702–1716. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.X.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef]
- Dixon, R.A.; Achnine, L.; Kota, P.; Liu, C.J.; Reddy, M.S.S.; Wang, L.J. The phenylpropanoid pathway and plant defence—A genomics perspective. Mol. Plant Pathol. 2002, 3, 371–390. [Google Scholar] [CrossRef]
- Hou, S.; Zhang, C.; Yang, Y.; Wu, D. Recent advances in plant immunity: Recognition, signaling, response, and evolution. Biol. Plant. 2013, 57, 11–25. [Google Scholar] [CrossRef]
- Zhu, H.; Yu, X.; Xu, T.; Wang, T.; Du, L.; Ren, G.; Dong, K. Transcriptome profiling of cold acclimation in bermudagrass (Cynodon dactylon). Sci. Hortic. 2015, 194, 230–236. [Google Scholar] [CrossRef]
- Kuang, X.; Sun, S.; Wei, J.; Li, Y.; Sun, C. Iso-Seq analysis of the Taxus cuspidata transcriptome reveals the complexity of Taxol biosynthesis. BMC Plant Biol. 2019, 19, 210. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; Cheng, X.; Luo, P.; Liang, F.; Wang, Z.; Yu, G.; Li, X.; Wang, D.; Bao, M. Hybrid sequencing and map finding (HySeMaFi): Optional strategies for extensively deciphering gene splicing and expression in organisms without reference genome. Sci. Rep. 2017, 7, 43793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichersky, E.; Raguso, R.A. Why do plants produce so many terpenoid compounds? New Phytol. 2018, 220, 692–702. [Google Scholar] [CrossRef]
- Martin, D.M.; Faldt, J.; Bohlmann, J. Functional characterization of nine Norway spruce TPS genes and evolution of gymnosperm terpene synthases of the TPS-d subfamily. Plant Physiol. 2004, 135, 1908–1927. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.S.; Xing, Z.; Liu, H.; Hu, X.G.; Gao, Q.; Xu, J.; Jiao, S.Q.; Jia, K.H.; Jin, Y.Q.; Zhao, W.; et al. In-depth transcriptome characterization uncovers distinct gene family expansions for Cupressus gigantea important to this long-lived species’ adaptability to environmental cues. BMC Genom. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Zhao, S.; Zhang, M.; Dong, K.; Chen, Y.; Fu, C.; Yu, L. Transcriptome assembly and systematic identification of novel cytochrome P450s in Taxus chinensis. Front. Plant Sci. 2017, 8, 1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, E.R. The evolution, function, structure, and expression of the plant sHSPs. J. Exp. Bot. 2013, 64, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, M.; Sun, Y.; Jia, C.; Peng, S.; Liu, Z.; Yang, G. Over-expression of JrsHSP17. 3 gene from Juglans regia confer the tolerance to abnormal temperature and NaCl stresses. J. Plant Biol. 2016, 59, 549–558. [Google Scholar] [CrossRef]
- Bonello, P.; Gordon, T.R.; Herms, D.A.; Wood, D.L.; Erbilgin, N. Nature and ecological implications of pathogen-induced systemic resistance in conifers: A novel hypothesis. Physiol. Mol. Plant Pathol. 2006, 68, 95–104. [Google Scholar] [CrossRef]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Robatzek, S.; Somssich, I.E. A new member of the Arabidopsis WRKY transcription factor family, AtWRKY6, is associated with both senescence- and defence-related processes. Plant J. 2001, 28, 123–133. [Google Scholar] [CrossRef]
- Jo, I.H.; Lee, J.; Hong, C.E.; Lee, D.J.; Bae, W.; Park, S.G.; Ahn, Y.J.; Kim, Y.C.; Kim, J.U.; Lee, J.W.; et al. Isoform sequencing provides a more comprehensive view of the Panax ginseng transcriptome. Genes 2017, 8, 228. [Google Scholar] [CrossRef] [Green Version]
- Hao, D.C.; Ge, G.B.; Xiao, P.G.; Zhang, Y.Y.; Yang, L. The first insight into the tissue specific Taxus transcriptome via Illumina second generation sequencing. PLoS ONE 2011, 6, e21220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fürtauer, L.; Weiszmann, J.; Weckwerth, W.; Nägele, T. Dynamics of plant metabolism during cold acclimation. Int. J. Mol. Sci. 2019, 20, 5411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Ding, G.; Cai, Q. The transcriptomic responses of Pinus massoniana to drought stress. Forests 2018, 9, 326. [Google Scholar] [CrossRef] [Green Version]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Kodaira, K.S.; Qin, F.; Tran, L.S.P.; Maruyama, K.; Kidokoro, S.; Fujita, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis Cys2/His2 zinc-finger proteins AZF1 and AZF2 negatively regulate abscisic acid-repressive and auxin-inducible genes under abiotic stress conditions. Plant Physiol. 2011, 157, 742–756. [Google Scholar] [CrossRef] [Green Version]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Kuijt, S.J.; Greco, R.; Agalou, A.; Shao, J.; CJ‘t Hoen, C.; Övernäs, E.; Osnato, M.; Curiale, S.; Meynard, D.; van Gulik, R.; et al. Interaction between the growth-regulating factor and knotted1-like homeobox families of transcription factors. Plant Physiol. 2014, 164, 1952–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Meng, X.; Dobrovolskaya, O.B.; Orlov, Y.L.; Chen, M. Non-coding RNAs and their roles in stress response in plants. Genom. Proteom. Bioinf. 2017, 15, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef]
- Xu, Q.; Song, Z.; Zhu, C.; Tao, C.; Kang, L.; Liu, W.; He, F.; Yan, J.; Sang, T. Systematic comparison of lncRNAs with protein coding mRNAs in population expression and their response to environmental change. BMC Plant Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Regulski, M.; Tseng, E.; Olson, A.; Goodwin, S.; McCombie, W.R.; Ware, D. A comparative transcriptional landscape of maize and sorghum obtained by single-molecule sequencing. Genome Res. 2018, 28, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef] [Green Version]
- Ben-Dov, C.; Hartmann, B.; Lundgren, J.; Valcarcel, J. Genome-wide analysis of alternative Pre-mRNA splicing. J. Biol. Chem. 2008, 283, 1229–1233. [Google Scholar] [CrossRef] [Green Version]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef]
- Reddy, A.S.N. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 2007, 58, 267–294. [Google Scholar] [CrossRef] [Green Version]
- Marquez, Y.; Brown, J.W.S.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatcher, S.R.; Zhou, W.; Leonard, A.; Wang, B.B.; Beatty, M.; Zastrow-Hayes, G.; Zhao, X.; Baumgarten, A.; Li, B. Genome-wide analysis of alternative splicing in Zea mays: Landscape and genetic regulation. Plant Cell 2014, 26, 3472–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.A.; Peng, D.L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, J.L.; Austin, M.; Stewart, C.J.; Noel, J. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakabayashi, R.; Mori, T.; Saito, K. Alternation of flavonoid accumulation under drought stress in Arabidopsis thaliana. Plant Signal. Behav. 2014, 9, e29518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Ye, X.; Li, J.; Tan, B.; Chen, P.; Cheng, J.; Wang, W.; Zheng, X.; Feng, J. Transcriptome profiling analysis revealed co-regulation of multiple pathways in jujube during infection by ‘Candidatus Phytoplasma ziziphi’. Gene 2018, 665, 82–95. [Google Scholar] [CrossRef]
- Keeling, C.I.; Bohlmann, J. Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defence of conifers against insects and pathogens. New Phytol. 2006, 170, 657–675. [Google Scholar] [CrossRef]
- Su, W.; Ye, C.; Zhang, Y.; Hao, S.; Li, Q.Q. Identification of putative key genes for coastal environments and cold adaptation in mangrove Kandelia obovata through transcriptome analysis. Sci. Total Environ. 2019, 681, 191–201. [Google Scholar] [CrossRef]
- Zhang, Y.; Nyong’A, T.M.; Shi, T.; Yang, P. The complexity of alternative splicing and landscape of tissue-specific expression in lotus (Nelumbo nucifera) unveiled by Illumina-and single-molecule real-time-based RNA-sequencing. DNA Res. 2019, 26, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jin, Y.; Huang, W.; Sun, Q.; Liu, F.; Huang, X. Full-length transcriptome sequences of ephemeral plant Arabidopsis pumila provides insight into gene expression dynamics during continuous salt stress. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef]
- Shi, Y.L.; Sheng, Y.Y.; Cai, Z.Y.; Yang, R.; Li, Q.S.; Li, X.M.; Li, D.; Guo, X.Y.; Lu, J.L.; Ye, J.H. Involvement of salicylic acid in anthracnose infection in tea plants revealed by transcriptome profiling. Int. J. Mol. Sci. 2019, 20, 2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharom, F.J.; Kretzschmar, T.; Burla, B.; Lee, Y.; Martinoia, E.; Nagy, R. Functions of ABC transporters in plants. Essays Biochem. 2011, 50, 145–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panikashvili, D.; Shi, J.X.; Bocobza, S.; Franke, R.B.; Schreiber, L.; Aharoni, A. The Arabidopsis DSO/ABCG11 transporter affects cutin metabolism in reproductive organs and suberin in roots. Mol. Plant 2010, 3, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Bostock, R.; Schaeffer, D.; Hammerschmidt, R. Comparison of elicitor activities of arachidonic acid, fatty acids and glucans from Phytophthora infestans in hypersensitivity expression in potato tuber. Physiol. Mol. Plant Pathol. 1986, 29, 349–360. [Google Scholar] [CrossRef]
- Li, H.; Li, M.; Wei, X.; Zhang, X.; Xue, R.; Zhao, Y.; Zhao, H. Transcriptome analysis of drought-responsive genes regulated by hydrogen sulfide in wheat (Triticum aestivum L.) leaves. Mol. Genet. Genom. 2017, 292, 1091–1110. [Google Scholar] [CrossRef] [PubMed]
- Aubourg, S.; Lecharny, A.; Bohlmann, J. Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol. Genet. Genom. 2002, 267, 730–745. [Google Scholar] [CrossRef]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.M.; Aubourg, S.; Schouwey, M.B.; Daviet, L.; Schalk, M.; Toub, O.; Lund, S.T.; Bohlmann, J. Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 2010, 10, 226. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.S.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015, 83, 189–212. [Google Scholar] [CrossRef]
- Hu, X.G.; Liu, H.; Jin, Y.; Sun, Y.Q.; Li, Y.; Zhao, W.; El-Kassaby, Y.A.; Wang, X.R.; Mao, J.F. De novo transcriptome assembly and characterization for the widespread and stress-tolerant conifer Platycladus orientalis. PLoS ONE 2016, 11, e0148985. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Keeling, C.I.; Weisshaar, S.; Ralph, S.G.; Jancsik, S.; Hamberger, B.; Dullat, H.K.; Bohlmann, J. Transcriptome mining, functional characterization, and phylogeny of a large terpene synthase gene family in spruce (Picea spp.). BMC Plant Biol. 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.; Madilao, L.L.; Ralph, S.; Bohlmann, J. Insect-induced conifer defense. White pine weevil and methyl jasmonate induce traumatic resinosis, de novo formed volatile emissions, and accumulation of terpenoid synthase and putative octadecanoid pathway transcripts in Sitka spruce. Plant Physiol. 2005, 137, 369–382. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.; Werck-Reichhart, D. A P450-centric view of plant evolution. Plant J. 2011, 66, 194–211. [Google Scholar] [CrossRef]
- Kim, J.A.; Roy, N.S.; Lee, I.H.; Choi, A.Y.; Choi, B.S.; Yu, Y.S.; Park, N.I.; Park, K.C.; Kim, S.; Yang, H.S.; et al. Genome-wide transcriptome profiling of the medicinal plant Zanthoxylum planispinum using a single-molecule direct RNA sequencing approach. Genomics 2019, 111, 973–979. [Google Scholar] [CrossRef]
- Kai, G.; Jiang, J.; Zhao, D.; Zhao, L.; Zhang, L.; Li, Z.; Guo, B.; Sun, X.; Miao, Z.; Tang, K. Isolation and expression profile analysis of a new cDNA encoding 5-alpha-taxadienol-10-beta-hydroxylase from Taxus media. J. Plant Biochem. Biotechnol. 2006, 15, 1–5. [Google Scholar] [CrossRef]
- Zhang, N.; Han, Z.; Sun, G.; Hoffman, A.; Wilson, I.W.; Yang, Y.; Gao, Q.; Wu, J.; Xie, D.; Dai, J.; et al. Molecular cloning and characterization of a cytochrome P450 taxoid 9a-hydroxylase in Ginkgo biloba cells. Biochem. Biophys. Res. Commun. 2014, 443, 938–943. [Google Scholar] [CrossRef]
- Ro, D.K.; Arimura, G.I.; Lau, S.Y.; Piers, E.; Bohlmann, J. Loblolly pine abietadienol/abietadienal oxidase PtAO (CYP720B1) is a multifunctional, multisubstrate cytochrome P450 monooxygenase. Proc. Natl. Acad. Sci. USA 2005, 102, 8060–8065. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.R. Plant cytochrome P450s from moss to poplar. Phytochem. Rev. 2006, 5, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Vasav, A.; Barvkar, V. Phylogenomic analysis of cytochrome P450 multigene family and their differential expression analysis in Solanum lycopersicum L. suggested tissue specific promoters. BMC Genom. 2019, 20, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, R.K.; De Domenico, S.; Santino, A. Plant cytochrome CYP74 family: Biochemical features, endocellular localisation, activation mechanism in plant defence and improvements for industrial applications. ChemBioChem 2009, 10, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Park, C.J.; Seo, Y.S. Heat shock proteins: A review of the molecular chaperones for plant immunity. Plant Pathol. J. 2015, 31, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Caitar, V.S.; de Carvalho, M.C.; Darben, L.M.; Kuwahara, M.K.; Nepomuceno, A.L.; Dias, W.P.; Abdelnoor, R.V.; Marcelino-Guimarães, F.C. Genome-wide analysis of the Hsp20 gene family in soybean: Comprehensive sequence, genomic organization and expression profile analysis under abiotic and biotic stresses. BMC Genom. 2013, 14, 577. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gao, T.; Wan, S.; Zhang, Y.; Yang, J.; Yu, Y.; Wang, W. Genome-wide identification, classification and expression analysis of the HSP gene superfamily in tea plant (Camellia sinensis). Int. J. Mol. Sci. 2018, 19, 2633. [Google Scholar] [CrossRef] [Green Version]
- Heath, L.S.; Ramakrishnan, N.; Sederoff, R.R.; Whetten, R.W.; Chevone, B.I.; Struble, C.A.; Jouenne, V.Y.; Chen, D.W.; van Zyl, L.; Grene, R. Studying the functional genomics of stress responses in loblolly pine with the Expresso microarray experiment management system. Comp. Funct. Genom. 2002, 3, 226–243. [Google Scholar] [CrossRef] [Green Version]
- Milioni, D.; Hatzopoulos, P. Genomic organization of hsp90 gene family in Arabidopsis. Plant Mol. Biol. 1997, 35, 955–961. [Google Scholar] [CrossRef]
- Krishna, P.; Gloor, G. The Hsp90 family of proteins in Arabidopsis thaliana. Cell Stress Chaperones 2001, 6, 238–246. [Google Scholar] [CrossRef]
- Liu, J.; Wang, R.; Liu, W.; Zhang, H.; Guo, Y.; Wen, R. Genome-wide characterization of heat-shock protein 70s from Chenopodium quinoa and expression analyses of Cqhsp70s in response to drought stress. Genes 2018, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, J.; Jia, H.; Yue, Z.; Lu, M.; Xin, X.; Hu, J. Genome-wide characterization of the sHsp gene family in Salix suchowensis reveals its functions under different abiotic stresses. Int. J. Mol. Sci. 2018, 19, 3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamara, M.; Parent, G.J.; Giguère, I.; Beaulieu, J.; Bousquet, J.; MacKay, J.J. Association genetics of acetophenone defence against spruce budworm in mature white spruce. BMC Plant Biol. 2018, 18, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossdal, C.G.; Sharma, P.; Lonneborg, A. Isolation of the first putative peroxidase cDNA from a conifer and the local and systemic accumulation of related proteins upon pathogen infection. Plant Mol. Biol. 2001, 47, 423–435. [Google Scholar] [CrossRef]
- Ralph, S.G.; Yueh, H.; Friedmann, M.; Aeschliman, D.; Zeznik, J.A.; Nelson, C.C.; Butterfield, Y.S.N.; Kirkpatrick, R.; Liu, J.; Jones, S.J.M.; et al. Conifer defence against insects: Microarray gene expression profiling of Sitka spruce (Picea sitchensis) induced by mechanical wounding or feeding by spruce budworms (Choristoneura occidentalis) or white pine weevils (Pissodes strobi) reveals large-scale changes of the host transcriptome. Plant Cell Environ. 2006, 29, 1545–1570. [Google Scholar]
- Hu, X.; Yang, J.; Li, C. Transcriptomic response to nitric oxide treatment in Larix olgensis Henry. Int. J. Mol. Sci. 2015, 16, 28582–28597. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.H.; Xu, L.L.; Tong, Z.K.; Lin, E.P.; Liu, Q.P.; Cheng, L.J.; Zhu, M.Y. De novo characterization of the Chinese fir (Cunninghamia lanceolata) transcriptome and analysis of candidate genes involved in cellulose and lignin biosynthesis. BMC Genom. 2012, 13, 648. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, H.; Mitsuhara, I.; Ito, N.; Seo, S.; Kamada, H.; Ohashi, Y. Transcriptionally and post-transcriptionally regulated response of 13 calmodulin genes to tobacco mosaic virus-induced cell death and wounding in tobacco plant. Eur. J. Biochem. 2001, 268, 3916–3929. [Google Scholar] [CrossRef]
- Yuenyong, W.; Chinpongpanich, A.; Comai, L.; Chadchawan, S.; Buaboocha, T. Downstream components of the calmodulin signaling pathway in the rice salt stress response revealed by transcriptome profiling and target identification. BMC Plant Biol. 2018, 18, 335. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Subramaniam, R.; Desveaux, D. Of guards, decoys, baits and traps: Pathogen perception in plants by type III effector sensors. Curr. Opin. Microbiol. 2016, 29, 49–55. [Google Scholar] [CrossRef]
- Chiang, Y.H.; Coaker, G. Effector triggered immunity: NLR immune perception and downstream defense responses. Arab. Book 2015, 2015, e0183. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J.; Staskawicz, B.J. Initiation of RPS2-specified disease resistance in Arabidopsis is coupled to the AvrRpt2-directed elimination of RIN4. Cell 2003, 112, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinf. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wu, T.D.; Reeder, J.; Lawrence, M.; Becker, G.; Brauer, M.J. GMAP and GSNAP for genomic sequence alignment: Enhancements to speed, accuracy, and functionality. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Springer: New York, NY, USA, 2016; Volume 1418, pp. 283–334. [Google Scholar]
- Alamancos, G.P.; Pages, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, U357–U359. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.N.; Li, B. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1.4.2: Tree Figure Drawing Tool. 2014. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 6 September 2019).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Root | Stem | Leaf | Strobilus |
|---|---|---|---|---|
| Nr | 38,641 | 45,531 | 46,770 | 53,725 |
| Swiss-Prot | 34,034 | 41,986 | 36,644 | 50,386 |
| KEGG | 37,452 | 43,929 | 46,196 | 51,357 |
| KOG | 26,123 | 31,452 | 26,955 | 38,723 |
| GO | 27,425 | 34,770 | 30,132 | 40,467 |
| Nt | 23,665 | 23,851 | 32,373 | 28,089 |
| Pfam | 27,425 | 34,770 | 30,132 | 40,467 |
| At least one database | 40,166 (89.46%) | 49,593 (85.38%) | 47,697 (94.48%) | 58,654 (86.72%) |
| All databases | 13,610 (30.31%) | 13,811 (23.78%) | 15,489 (30.68%) | 16,641 (24.60%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Wang, Z.; Su, Y.; Wang, T. Characterization and Analysis of the Full-Length Transcriptomes of Multiple Organs in Pseudotaxus chienii (W.C.Cheng) W.C.Cheng. Int. J. Mol. Sci. 2020, 21, 4305. https://doi.org/10.3390/ijms21124305
Liu L, Wang Z, Su Y, Wang T. Characterization and Analysis of the Full-Length Transcriptomes of Multiple Organs in Pseudotaxus chienii (W.C.Cheng) W.C.Cheng. International Journal of Molecular Sciences. 2020; 21(12):4305. https://doi.org/10.3390/ijms21124305
Chicago/Turabian StyleLiu, Li, Zhen Wang, Yingjuan Su, and Ting Wang. 2020. "Characterization and Analysis of the Full-Length Transcriptomes of Multiple Organs in Pseudotaxus chienii (W.C.Cheng) W.C.Cheng" International Journal of Molecular Sciences 21, no. 12: 4305. https://doi.org/10.3390/ijms21124305
APA StyleLiu, L., Wang, Z., Su, Y., & Wang, T. (2020). Characterization and Analysis of the Full-Length Transcriptomes of Multiple Organs in Pseudotaxus chienii (W.C.Cheng) W.C.Cheng. International Journal of Molecular Sciences, 21(12), 4305. https://doi.org/10.3390/ijms21124305