Optimization of a WGA-Free Molecular Tagging-Based NGS Protocol for CTCs Mutational Profiling

 ,
, 


Abstract
:1. Introduction
2. Results
2.1. DEPArrayTM Sorting and Validation of Optimized NGS Procedures
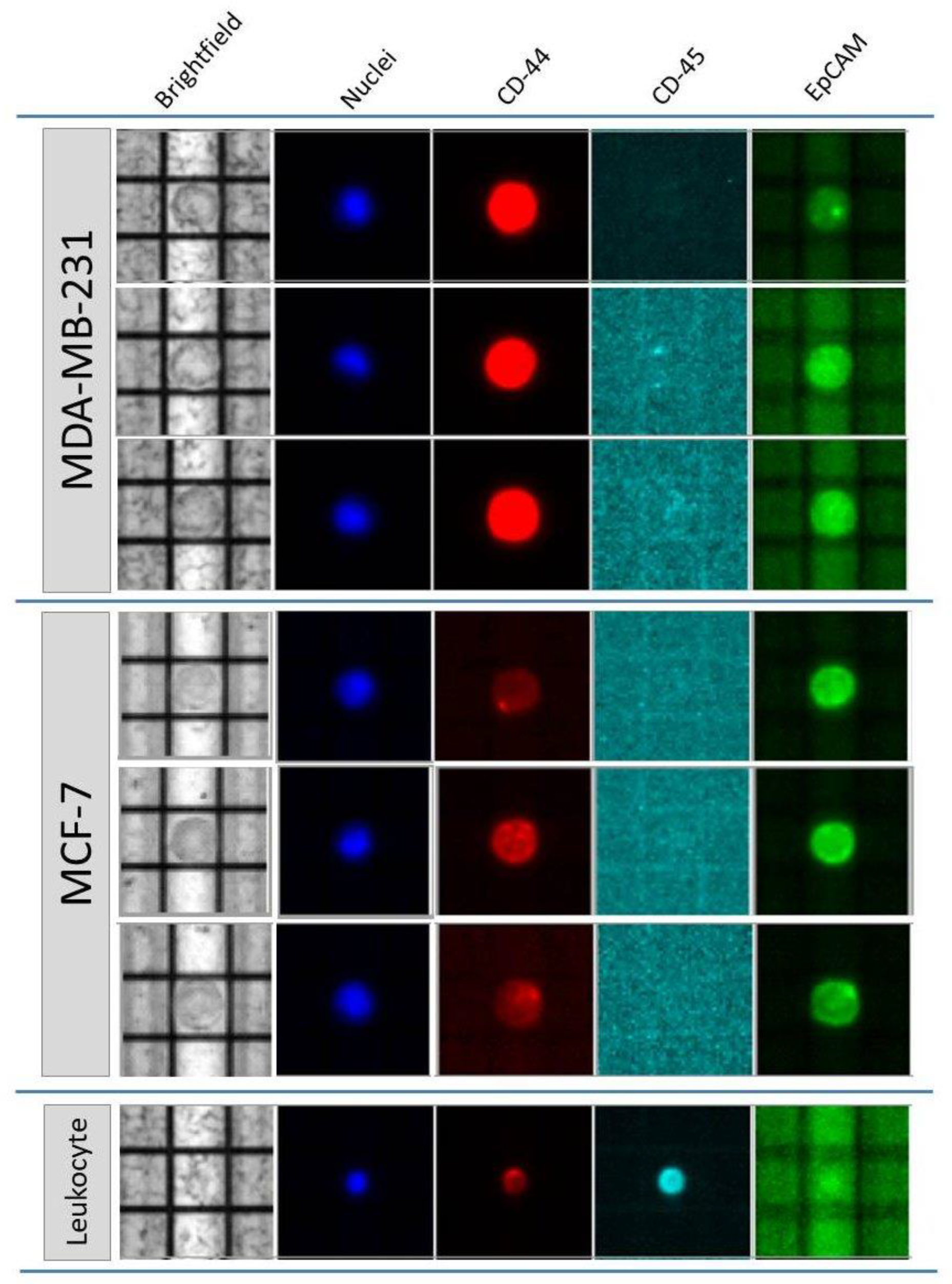
2.1.1. Cell Detection and DNA Integrity Assessment
2.1.2. From Canonical to Optimized Molecular Tagging NGS Workflow: Experimental Setting and Development
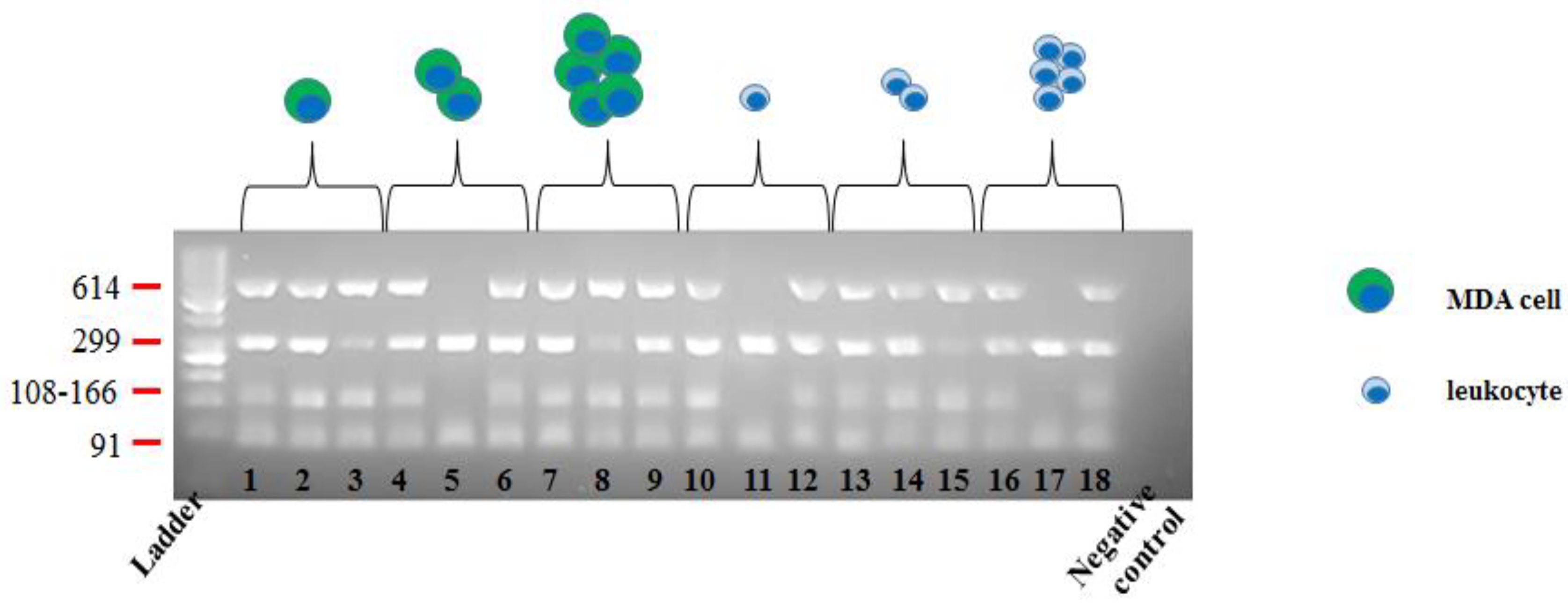
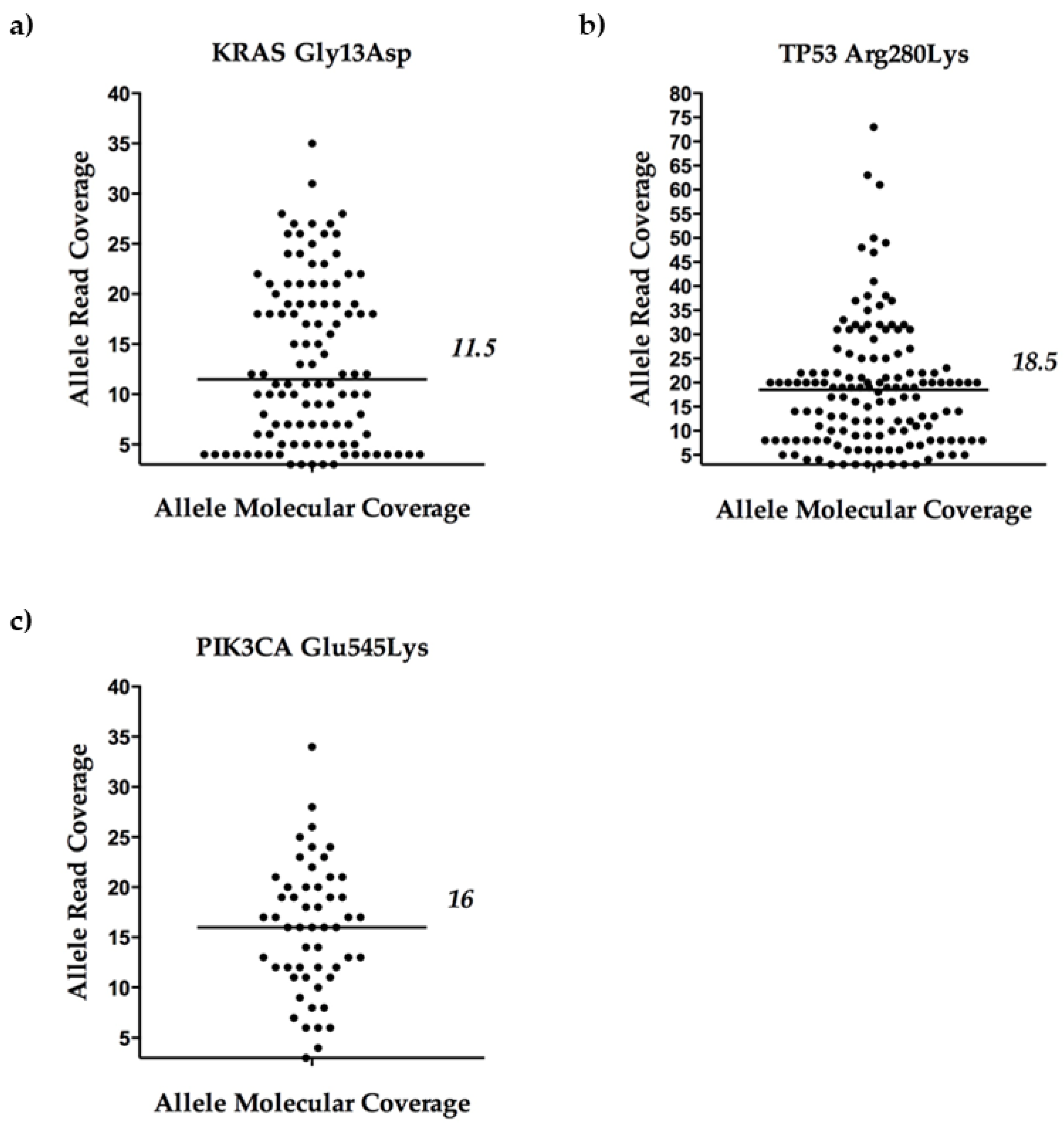
2.1.3. A glance at Variant Calling and Coverage Metrics
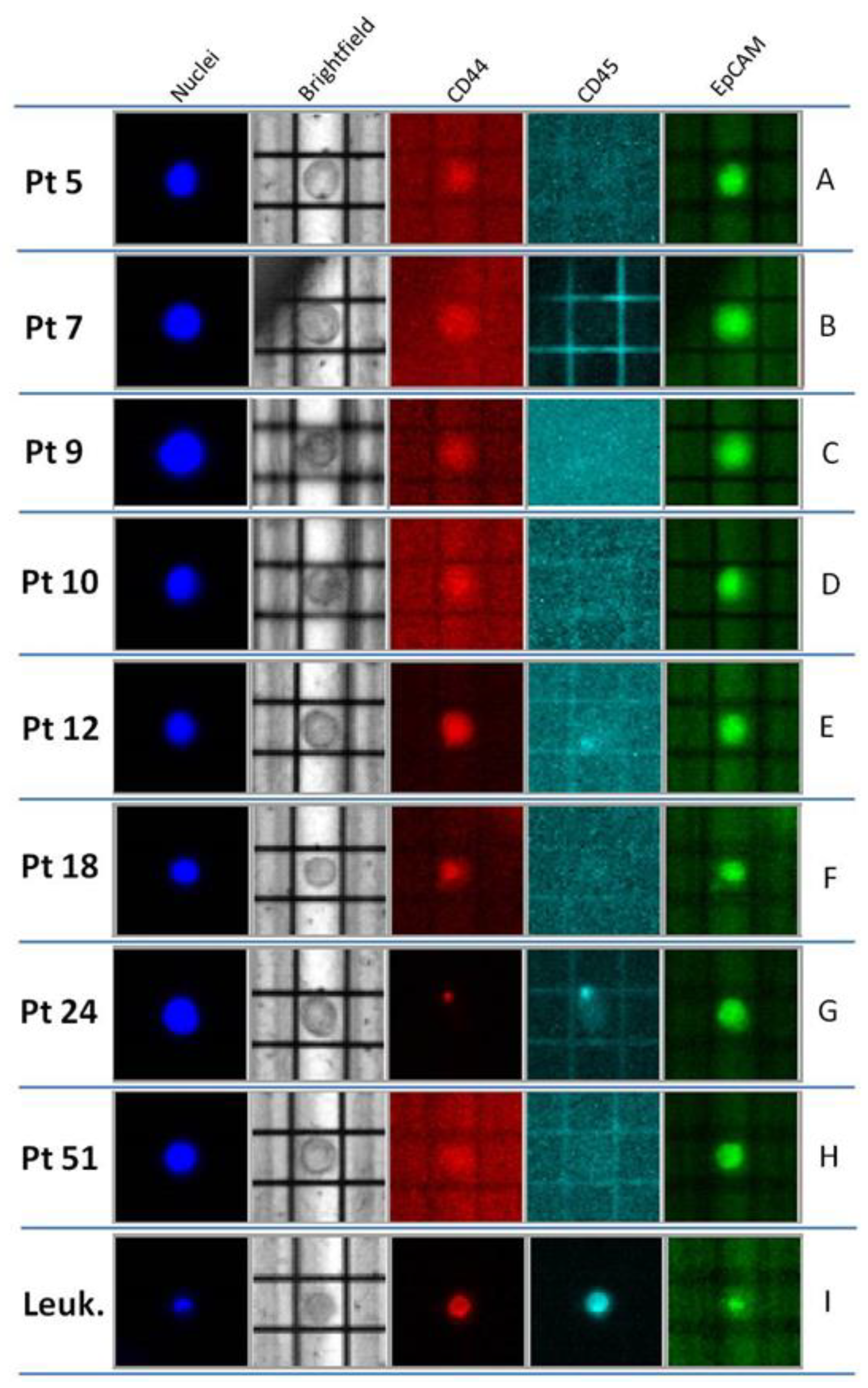
2.2. Optimized Molecular Tagging NGS Workflow Revealed to be Reliable for Molecular Analysis of CTCs
3. Discussion
4. Materials and Methods
4.1. Method Optimization
4.1.1. Cell Line, Spiking Experiments, and DEPArrayTM Cell Sorting
4.1.2. Evaluation of DNA Quality
4.1.3. Canonical NGS Workflow Assessment
4.1.4. Optimized Molecular Tagging NGS Workflow
4.1.5. NGS Analysis
4.2. CTCs Testing
4.2.1. BC Patients
4.2.2. Enrichment, Fixation, and Sorting of CTCs from BC Patients
4.3. NGS of BC Tissues
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CTCs | Circulating Tumor Cells |
| WGA | Whole Genome Amplification |
| NGS | Next-Generation Sequencing |
| ctDNA | Circulating tumor DNA |
| BC | Breast Cancer |
| MDA | MDA-MB-231 |
| EpCAM | Epithelial Cell Adhesion Molecule |
| FITC | fluorescein isothiocyanate |
| PE | phycoerythrin |
| APC | allophycocyanin |
| FACS | Fluorescent-Activated Cell Sorter |
| PCR | Polymerase Chain Reaction |
| OBcfRAv2 | Oncomine Breast cfDNA Research Assay v2 |
| KRAS | Kirsten rat sarcoma 2 viral oncogene homolog |
| TP53 | Tumor Protein 53 |
| MAFs | Molecular Allele Frequencies |
| VFSH | Variant Family Size Histogram |
| TVC | Torrent Variant Caller |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| ERBB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| ERBB3 | Erb-B2 Receptor Tyrosine Kinase 3 |
| ERBB4 | Erb-B2 Receptor Tyrosine Kinase 4 |
| ESR1 | Estrogen Receptor 1 |
| MCL1 | MCL1 Apoptosis Regulator |
| GATA3 | GATA Binding Protein 3 |
| PTEN | Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase |
| CCND1 | Cyclin D1 |
| AKT1 | v-akt murine thymoma viral oncogene homolog 1 |
| CDH1 | Cadherin 1 |
| MAP2K4 | Mitogen-Activated Protein Kinase Kinase 4 |
| SF3B1 | Splicing factor 3b subunit 1 |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| MAP3K1 | Mitogen-Activated Protein Kinase Kinase Kinase 1 |
| PIK3R1 | Phosphoinositide-3-Kinase Regulatory Subunit 1 |
| EGFR | Epidermal Growth Factor Receptor |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| BAM | Binary Alignment Map |
| VCF | Variant Caller Format |
| IGV | Integrative Genomic Viewer |
| TMAP | Torrent Mapping Alignment Program |
| DMEM | Dulbecco’s Modified Eagle Medium |
| PBS | Phosphate Buffered Saline |
| BEAMing | Beads, Emulsions, Amplification, and Magnetics |
References
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization. J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Peeters, D.J.; Fehm, T.; Nolé, F.; Criado, R.G.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Huang, X.; Gao, P.; Song, Y.; Sun, J.; Chen, X.; Zhao, J.; Xu, H.; Wang, Z. Meta-analysis of the prognostic value of circulating tumor cells detected with the CellSearch System in colorectal cancer. BMC Cancer 2015, 15, 202. [Google Scholar] [CrossRef] [Green Version]
- Tol, J.; Koopman, M.; Miller, M.C.; Tibbe, A.; Cats, A.; Creemers, G.J.M.; Vos, A.H.; Nagtegaal, I.D.; Terstappen, L.W.M.M.; Punt, C.J.A. Circulating tumour cells early predict progression-free and overall survival in advanced colorectal cancer patients treated with chemotherapy and targeted agents. Ann. Oncol. 2010, 21, 1006–1012. [Google Scholar] [CrossRef]
- Krebs, M.G.; Sloane, R.; Priest, L.; Lancashire, L.; Hou, J.M.; Greystoke, A.; Ward, T.H.; Ferraldeschi, R.; Hughes, A.; Clack, G.; et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 1556–1563. [Google Scholar] [CrossRef]
- Sparano, J.; O’Neill, A.; Alpaugh, K.; Wolff, A.C.; Northfelt, D.W.; Dang, C.T.; Sledge, G.W.; Miller, K.D. Association of Circulating Tumor Cells with Late Recurrence of Estrogen Receptor-Positive Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1700–1706. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Moon, D.H.; Lindsay, D.P.; Hong, S.; Wang, A.Z. Clinical indications for, and the future of, circulating tumor cells. Adv. Drug Deliv. Rev. 2018, 125, 143–150. [Google Scholar] [CrossRef]
- Lianidou, E.; Pantel, K. Liquid Biopsies. Genes Chromosomes Cancer 2019, 58, 219–232. [Google Scholar] [CrossRef]
- Zhang, H.; Gong, S.; Liu, Y.; Liang, L.; He, S.; Zhang, Q.; Si, M.; Yu, Z. Enumeration and molecular characterization of circulating tumor cell using an in vivo capture system in squamous cell carcinoma of head and neck. Chin. J. Cancer Res. 2017, 29, 196–203. [Google Scholar] [CrossRef]
- Thery, L.; Meddis, A.; Cabel, L.; Proudhon, C.; Latouche, A.; Pierga, J.Y.; Bidard, F.C. Circulating Tumor Cells in Early Breast Cancer. JNCI Cancer Spectrum. 2019, 3, pkz026. [Google Scholar] [CrossRef] [PubMed]
- Salvianti, F.; Rotunno, G.; Galardi, F.; De Luca, F.; Pestrin, M.; Vannucchi, A.M.; Di Leo, A.; Pazzagli, M.; Pinzani, P. Feasibility of a workflow for the molecular characterization of single cells by next generation sequencing. Biomol. Detect. Quantif. 2015, 5, 23–29. [Google Scholar] [CrossRef] [Green Version]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.S.; Lieberman, D.B.; Blanchard, T.; Rader, J.; Zhao, J.; Troxel, A.B.; DeSloover, D.; Fox, A.J.; Daber, R.D.; Kakrecha, B. A novel approach for next-generation sequencing of circulating tumor cells. Mol. Genet. Genom. Med. 2016, 4, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sho, S.; Court, C.M.; Winograd, P.; Lee, S.; Hou, S.; Graeber, T.G.; Tseng, H.R.; Tomlinson, J.S. Precision oncology using a limited number of cells: Optimization of whole genome amplification products for sequencing applications. BMC Cancer 2017, 17, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinard, R.; de Winter, A.; Sarkis, G.J.; Gerstein, M.B.; Tartaro, K.R.; Plant, R.N.; Egholm, M.; Rothberg, J.M.; Leamon, J.H. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genom. 2006, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; Egholm, M. Whole genome amplification: Abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol. 2003, 21, 531–535. [Google Scholar] [CrossRef]
- Hughes, S.; Arneson, N.; Done, S.; Squire, J. The use of whole genome amplification in the study of human disease. Prog. Biophys. Mol. Biol. 2005, 88, 173–189. [Google Scholar] [CrossRef]
- Huang, L.; Ma, F.; Chapman, A.; Lu, S.; Xie, X.S. Single-cell whole-genome amplification and sequencing: Methodology and applications. Annu. Rev. Genom. Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Qiu, S.; Shao, K.; Hou, Y. Progress and challenges of sequencing and analyzing circulating tumor cells. Cell Biol. Toxicol. 2018, 34, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahn, H.; Steif, A.; Laks, E.; Eirew, P.; VanInsberghe, M.; Shah, S.P.; Aparicio, S.; Hansen, C.L. Scalable whole-genome single-cell library preparation without preamplification. Nat. Methods 2017, 14, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Palmirotta, R.; Lovero, D.; Silvestris, E.; Felici, C.; Quaresmini, D.; Cafforio, P.; & Silvestris, F. Next-generation Sequencing (NGS) Analysis on Single Circulating Tumor Cells (CTCs) with No Need of Whole-genome Amplification (WGA). Cancer Genom. Proteom. 2017, 14, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Oronzo, S.; Lovero, D.; Palmirotta, R.; Stucci, L.S.; Tucci, M.; Felici, C.; Cascardi, E.; Giardina, C.; Caforio, P.; Silvestris, F. Dissection of major cancer gene variants in subsets of circulating tumor cells in advanced breast cancer. Sci. Rep. 2019, 9, 17276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W.W.M. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res. 2006, 12, 4218–4224. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, N.; Hu, P.; Bedard, P.; Clemons, M.; McCready, D.; Done, S.J. Identification of genomic signatures in circulating tumor cells from breast cancer. Int. J. Cancer 2015, 137, 332–344. [Google Scholar] [CrossRef]
- Liu, H.E.; Triboulet, M.; Zia, A.; Vuppalapaty, M.; Kidess-Sigal, E.; Coller, J.; Natu, V.S.; Shokoohi, V.; Che, J.; Renier, C.; et al. Workflow optimization of whole genome amplification and targeted panel sequencing for CTC mutation detection. NPJ Genom. Med. 2017, 2, 34. [Google Scholar] [CrossRef]
- Bingham, C.; Fernandez, S.V.; Fittipaldi, P.; Dempsey, P.W.; Ruth, K.J.; Cristofanilli, M.; Katherine Alpaugh, R. Mutational studies on single circulating tumor cells isolated from the blood of inflammatory breast cancer patients. Breast Cancer Res. Treat. 2017, 163, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Dono, M.; De Luca, G.; Lastraioli, S.; Anselmi, G.; Dal Bello, M.G.; Coco, S.; Vanni, I.; Grossi, F.; Vigani, A.; Genova, C.; et al. Tag-based next generation sequencing: A feasible and reliable assay for EGFR T790M mutation detection in circulating tumor DNA of non small cell lung cancer patients. Mol. Med. 2019, 25, 15. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Lastraioli, S.; Conte, R.; Mora, M.; Genova, C.; Rossi, G.; Tagliamento, M.; Coco, S.; Dal Bello, M.G.; Zupo, S.; et al. Performance of the OncomineTM Lung cfDNA Assay for Liquid Biopsy by NGS of NSCLC Patients in Routine Laboratory Practice. Appl. Sci. 2020, 10, 2895. [Google Scholar] [CrossRef]
- Abaan, O.D.; Polley, E.C.; Davis, S.R.; Zhu, Y.J.; Bilke, S.; Walker, R.L.; Pineda, M.; Gindin, Y.; Jiang, Y.; Reinhold, W.C. The exomes of the NCI-60 panel: A genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013, 73, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Hollestelle, A.; Nagel, J.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.; French, P.; Peeters, J.; Rozendaal, M. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Vijaya Satya, R.; Lewis, M.; Randad, P.; Wang, Y. Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genom. 2015, 7, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. SOLAR-1 Study Group. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Pestrin, M.; Salvianti, F.; Galardi, F.; De Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; Di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, S.V.; Bingham, C.; Fittipaldi, P.; Austin, L.; Palazzo, J.; Palmer, G.; Alpaugh, K.; Cristofanilli, M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res. 2014, 16, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolillo, C.; Mu, Z.; Rossi, G.; Schiewer, M.J.; Nguyen, T.; Austin, L.; Capoluongo, E.; Knudsen, K.; Cristofanilli, M.; Fortina, P. Detection of Activating Estrogen Receptor Gene (ESR1) Mutations in Single Circulating Tumor Cells. Clin. Cancer Res. 2017, 23, 6086–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Foncillas, J.; Alba, E.; Aranda, E.; Díaz-Rubio, E.; López-López, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Bulfoni, M.; Gerratana, L.; Del Ben, F.; Marzinotto, S.; Sorrentino, M.; Turetta, M.; Scoles, G.; Toffoletto, B.; Isola, M.; Beltrami, C.A.; et al. In patients with metastatic breast cancer the identification of circulating tumor cells in epithelial-to-mesenchymal transition is associated with a poor prognosis. Breast Cancer Res. 2016, 18, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
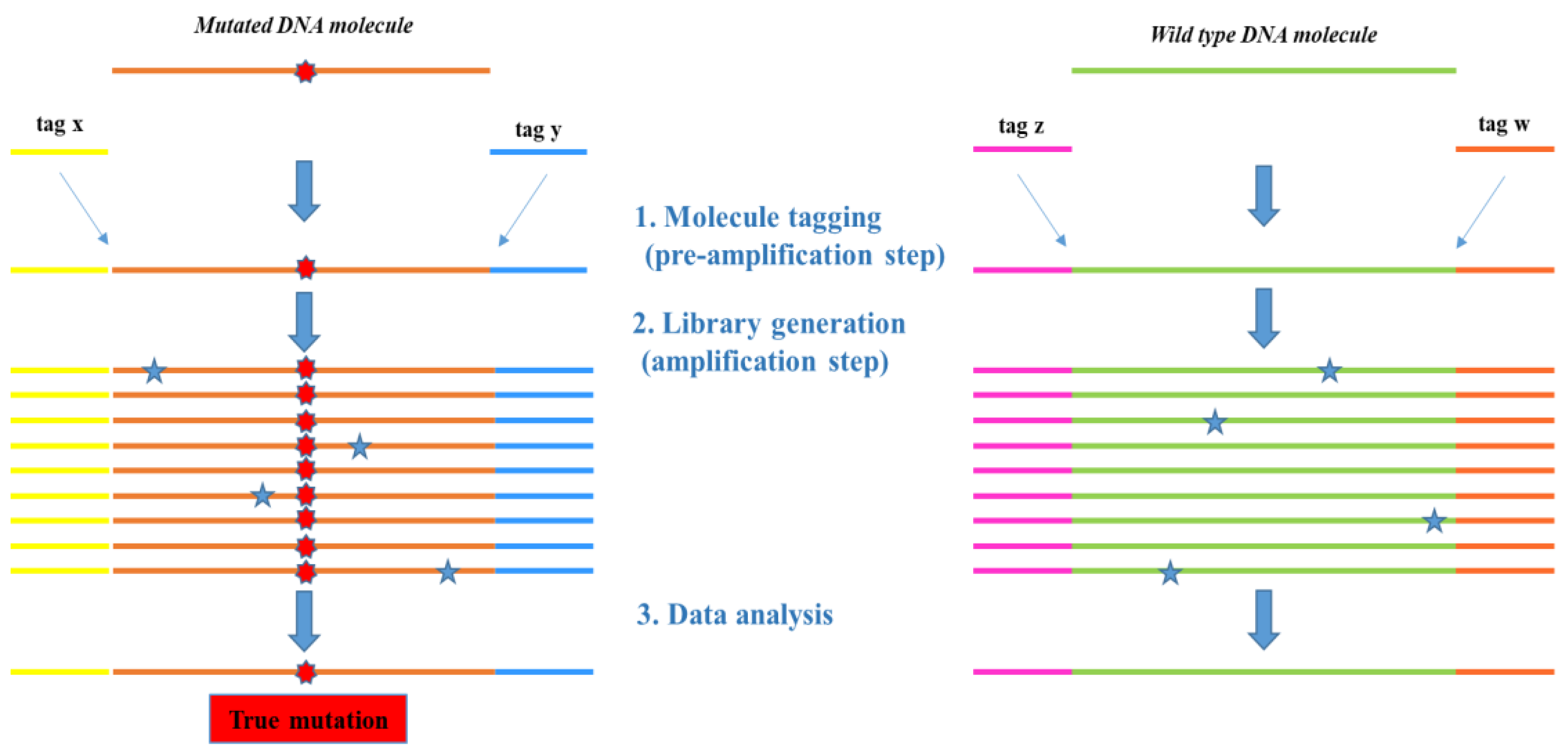{kind=link}
| Canonical Molecular Tagging NGS Workflow | Optimized Molecular Tagging NGS Workflow | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Types of starting isolated cells | MDA cells (single or pools) | MDA a/MCF-7 b cells (only pools) | MDA/MCF-7: leukocytes (combined pools) | Leukocytes (only pools) | ||||||||
| n of cells | 1 | 3 | 5 | 2 | 4 | 5 | 4:1 | 3:2 | 2:3 | 1:4 | 2 | 5 |
| 5ng of DNA from healthy donor leukocytes | + | + | + | − | − | − | − | − | − | − | − | − |
| OBcfRAv2 volume reagents reaction | As recommended by manufacturer | Modified: 3× reduction of volume reagents | ||||||||||
| Thermal PCR conditions | As recommended by manufacturer | As recommended by manufacturer | ||||||||||
| Libraries quantification | qPCR | qPCR | ||||||||||
| Libraries multiplexing on chip | Five libraries on Ion 530TM Chip | 24 libraries on Ion 520TM Chip (low-coverage sequencing) | ||||||||||
| MDA-MB-231 | MCF-7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| KRAS Gly13Asp | TP53 Arg280Lys | PIK3CA Glu545Lys | ||||||||
| Pool Type | n of Cells | Expected MAFs (%) | Mean Observed MAFs (%) | Median Allele Molecular Coverage | Expected MAFs (%) | Mean Observed MAFs (%) | Median Allele Molecular Coverage | Expected MAFs (%) | Mean Observed MAFs (%) | Median Allele Molecular Coverage |
| tumor cells | 2 | 100 | 100 | 7 | 100 | 100 | 10 | 50 | 38 | 2 |
| 4 | 100 | 100 | 8 | 100 | 100 | 12 | 50 | 66.7 | 2.5 | |
| 5 | 100 | 100 | 10 | 100 | 95.8 | 14 | 50 | 43.7 | 6.5 | |
| tumor cells: leukocytes | 4:1 | 80 | 76.2 | 5 | 80 | 87.5 | 7 | 40 | 33.3 | 6 |
| 3:2 | 60 | 83.4 a | 2 a | 60 | 57.5 a | 6 a | 30 | 50 | 3 | |
| 2:3 | 40 | 37.2 | 5 | 40 | 57.6 | 5 | 20 | 36.4 b | 4 b | |
| 1:4 | 20 | 24.4 | 2 | 20 | 45.1 | 3 | 10 | 14.8 | 1 | |
| Leukocytes | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Pt | CTCs | Concordance CTC/Tissue | BC Tissue Mutations | ||
|---|---|---|---|---|---|
| (Number Cells/Reaction) | Mutations (OBcfRAv2) | Detectable by OBcfRAv2 and Custom Panel | Detectable Only by Custom Panel | ||
| 5 | 3 | TP53 c.1100 + 30A > T | yes | TP53 c.1100 + 30A > T | ERBB2 p.Ile654Val ERBB2 p.Ile655Val ERBB2 p.Pro1170Ala |
| 7 | 3 | TP53 p.Arg213= | yes | TP53 p.Arg213= | PIK3CA p.Asn345Lys ERBB2 p.Ile655Val |
| 9 | 3 | not found | yes | not found | PIK3R1 p.Met326Ile ERBB2 p.Pro1170Ala |
| 10 | 2 | not found | no | TP53 p.Arg248Gln | |
| 2 | not found | ||||
| 4 | not found | ||||
| 12 | 5 | TP53 c.1100 + 30A > T | yes | TP53 c.1100 + 30A > T | ERBB2 p.Pro1170Ala |
| 18 | 3 | not found | no | TP53 p.Cys275Leufs | ERBB2 p.Pro1170Ala |
| 3 | not found | ||||
| 24 | 2 | TP53 c.1100 + 30A > T | partial | TP53 p.Arg248Trp TP53 c.1100 + 30A > T | ERBB2 p.Pro1170Ala |
| 5 | TP53 c.1100 + 30A > T | ||||
| 51 | 3 | PIK3CA p. Hys1047Arg | yes | PIK3CA p. Hys1047Arg | |
| 6 | PIK3CA p. Hys1047Arg | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, G.; Cardinali, B.; Del Mastro, L.; Lastraioli, S.; Carli, F.; Ferrarini, M.; Calin, G.A.; Garuti, A.; Mazzitelli, C.; Zupo, S.; et al. Optimization of a WGA-Free Molecular Tagging-Based NGS Protocol for CTCs Mutational Profiling. Int. J. Mol. Sci. 2020, 21, 4364. https://doi.org/10.3390/ijms21124364
De Luca G, Cardinali B, Del Mastro L, Lastraioli S, Carli F, Ferrarini M, Calin GA, Garuti A, Mazzitelli C, Zupo S, et al. Optimization of a WGA-Free Molecular Tagging-Based NGS Protocol for CTCs Mutational Profiling. International Journal of Molecular Sciences. 2020; 21(12):4364. https://doi.org/10.3390/ijms21124364
Chicago/Turabian StyleDe Luca, Giuseppa, Barbara Cardinali, Lucia Del Mastro, Sonia Lastraioli, Franca Carli, Manlio Ferrarini, George A. Calin, Anna Garuti, Carlotta Mazzitelli, Simona Zupo, and et al. 2020. "Optimization of a WGA-Free Molecular Tagging-Based NGS Protocol for CTCs Mutational Profiling" International Journal of Molecular Sciences 21, no. 12: 4364. https://doi.org/10.3390/ijms21124364
APA StyleDe Luca, G., Cardinali, B., Del Mastro, L., Lastraioli, S., Carli, F., Ferrarini, M., Calin, G. A., Garuti, A., Mazzitelli, C., Zupo, S., & Dono, M. (2020). Optimization of a WGA-Free Molecular Tagging-Based NGS Protocol for CTCs Mutational Profiling. International Journal of Molecular Sciences, 21(12), 4364. https://doi.org/10.3390/ijms21124364