The Impact of Matrix Metalloproteinase-9 on the Sequential Steps of the Metastatic Process

Abstract
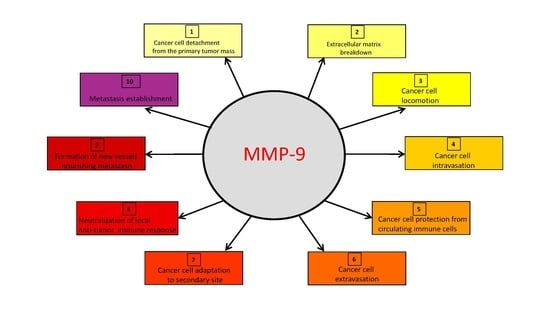
:
1. Introduction
2. MMP-9 Production or Activity is Regulated at Multiple Levels
3. MMP-9 Favors Cancer Cell Detachment from the Primary Tumor by Loosening their Adhesive Interactions and Enhancing their Invasive Capabilities
4. MMP-9 Aids Cancer Cell Trafficking Inside and Outside the Lymphatic or Blood Vessels
5. MMP-9 Favors Tumor Cell Adaptation to the Microenvironment of the Secondary Site, Hence Effectively Participating in the Establishment of Metastasis
6. MMP-9 Inhibitors
7. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | protein kinase B |
| CXCL | CXC chemokine ligand |
| CXCR | CXC chemokine receptor |
| ECM | extracellular matrix |
| EMT | epithelial-to-mesenchymal transition |
| ERK | extracellular signal-regulated kinase |
| HIF | hypoxia inducible factor |
| HIV | human immunodeficiency virus |
| MAPK | mitogen activated protein kinase |
| MET | mesenchymal-to-epithelial transition |
| MMP | matrix metalloproteinase |
| MT-MMP | membrane type-matrix metalloproteinase |
| NF-kB | nuclear factor-kappa B |
| PI3K | phosphoinositide 3 kinase |
| PIP3 | phosphatidylinositol-3,4,5-trisphosphate |
| TIMP | tissue inhibitor of matrix metalloproteinase |
| VEGF | vascular endothelial growth factor |
References
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Høye, A.; Erler, J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Physiol. 2016, 310, C955–C967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Membrane-Type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2036–2042. [Google Scholar] [CrossRef]
- Stanciu, A.E.; Zamfir-Chiru-Anton, A.; Stanciu, M.; Popescu, C.; Gheorghe, D. Imbalance between Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases Promotes Invasion and Metastasis of Head and Neck Squamous Cell Carcinoma. Clin. Lab. 2017, 63, 1613–1620. [Google Scholar] [CrossRef]
- Yao, Q.; Kou, L.; Tu, Y.; Zhu, L. MMP-Responsive ‘Smart’ Drug Delivery and Tumor Targeting. Trends Pharmacol. Sci. 2018, 39, 766–781. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [Green Version]
- Hurt, B.; Schulick, R.; Edil, B.; El Kasmi, K.C.; Barnett, C. Cancer-Promoting mechanisms of tumor-Associated neutrophils. Am. J. Surg. 2017, 214, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Walia, A.; Yang, J.; Huang, Y.-H.; Rosenblatt, M.I.; Chang, J.-H.; Azar, D.T. Endostatin’s emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim. Biophys. Acta 2015, 1850, 2422–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Fouad, H.; Salem, H.; Ellakwa, D.E.-S.; Abdel-Hamid, M. MMP-2 and MMP-9 as prognostic markers for the early detection of urinary bladder cancer. J. Biochem. Mol. Toxicol. 2018, 33, e22275. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Dai, Y.; Wang, A.; Wang, C.; Sun, L.; Wang, Z. Association between expression of MMP-7 and MMP-9 and pelvic lymph node and para-Aortic lymph node metastasis in early cervical cancer. J. Obstet. Gynaecol. Res. 2018, 44, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Dagher, A.; Sachdev, M.; Ebi, M.; Yamada, T.; Joh, T.; Moses, M. Urinary ADAM12 and MMP-9/NGAL Complex Detect the Presence of Gastric Cancer. Cancer Prev. Res. 2015, 8, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Newby, A. Metalloproteinase production from macrophages—Perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 2016, 101, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Wang, Y.; Liu, Z.; Wang, L.; Cao, Z.; Zhang, C.; Hao, Y.; He, H. Effects of IL-1β on MMP-9 Expression in Cementoblast-Derived Cell Line and MMP-Mediated Degradation of Type I Collagen. Inflammation 2019, 42, 413–425. [Google Scholar] [CrossRef]
- Ding, X.-W.; Sun, X.; Shen, X.-F.; Lu, Y.; Wang, J.-Q.; Sun, Z.-R.; Miao, C.-H.; Chen, J. Propofol attenuates TNF-α-induced MMP-9 expression in human cerebral microvascular endothelial cells by inhibiting Ca2+/CAMK II/ERK/NF-κB signaling pathway. Acta Pharmacol. Sin. 2019, 40, 1303–1313. [Google Scholar] [CrossRef]
- Barillari, G.; Iovane, A.; Bacigalupo, I.; Palladino, C.; Bellino, S.; Leone, P.; Monini, P.; Ensoli, B. Ritonavir or saquinavir impairs the invasion of cervical intraepithelial neoplasia cells via a reduction of MMP expression and activity. AIDS 2012, 26, 909–919. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Lee, Y.-C.; Huang, C.-H.; Chang, L.-S. Gallic acid-capped gold nanoparticles inhibit EGF-induced MMP-9 expression through suppression of p300 stabilization and NFκB/c-Jun activation in breast cancer MDA-MB-231 cells. Toxicol. Appl. Pharmacol. 2016, 310, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Vetrugno, C.; Cossa, L.G.; Marsigliante, S. TGF-β1 activates RSC96 Schwann cells migration and invasion through MMP-2 and MMP-9 activities. J. Neurochem. 2019, 153, 525–538. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Tsai, H.-L.; Syu, J.-S.; Chen, T.-Y.; Su, M.-T. Primary Cilium-Regulated EG-VEGF Signaling Facilitates Trophoblast Invasion. J. Cell. Physiol. 2016, 232, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zheng, M.-Q.; Lu, J.-W.; Jiang, Q.; Wang, T.-H.; Huang, X.-E. CXCL12-CXCR4 Promotes Proliferation and Invasion of Pancreatic Cancer Cells. Asian Pac. J. Cancer Prev. 2013, 14, 5403–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravani, R.; Yari, D.; Hasanian-Langroudi, F. Correlation between the COL4A3, MMP-9, and TIMP-1 polymorphisms and risk of keratoconus. Jpn. J. Ophthalmol. 2017, 61, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Veidal, S.S.; Larsen, D.; Chen, X.; Sun, S.; Zheng, Q.; Bay-Jensen, A.-C.; Leeming, D.; Nawrocki, A.; Larsen, M.R.; Schett, G.; et al. MMP mediated type V collagen degradation (C5M) is elevated in ankylosing spondylitis. Clin. Biochem. 2012, 45, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Van Doren, S.R. Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol. 2015, 44, 224–231. [Google Scholar] [CrossRef]
- Li, W.; Liu, Z.; Zhao, C.; Zhai, L. Binding of MMP-9-Degraded fibronectin to ?6 integrin promotes invasion via the FAK-Src-Related Erk1/2 and PI3K/Akt/Smad-1/5/8 pathways in breast cancer. Oncol. Rep. 2015, 34, 1345–1352. [Google Scholar] [CrossRef]
- Darweish, M.M.; Abbas, A.; Ebrahim, M.A.; Al-Gayyar, M.H. Chemopreventive and hepatoprotective effects of Epigallocatechin-Gallate against hepatocellular carcinoma: Role of heparan sulfate proteoglycans pathway. J. Pharm. Pharmacol. 2014, 66, 1032–1045. [Google Scholar] [CrossRef]
- Van den Steen, P.E.; Husson, S.J.; Proost, P.; Van Damme, J.; Opdenakker, G. Carboxyterminal cleavage of the chemokines MIG and IP-10 by gelatinase B and neutrophil collagenase. Biochem. Biophys. Res. Commun. 2003, 310, 889–896. [Google Scholar] [CrossRef]
- Sheu, B.C.; Hsu, S.M.; Ho, H.N.; Lien, H.C.; Huang, S.C.; Lin, R.H. A novel role of metalloproteinase in cancer-Mediated immunosuppression. Cancer Res. 2001, 61, 237–242. [Google Scholar] [PubMed]
- Philipp, K.; Riedel, F.; Sauerbier, M.; Hormann, K.; Germann, G. Targeting TGF-Beta in human keratinocytes and its potential role in wound healing. Int. J. Mol. Med. 2004, 14, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Van Der Kooij, M.A.; Fantin, M.; Rejmak-Kozicka, E.; Grosse, J.; Zanoletti, O.; Fournier, C.; Ganguly, K.; Kalita, K.; Kaczmarek, L.; Sandi, C. Role for MMP-9 in stress-Induced downregulation of nectin-3 in hippocampal CA1 and associated behavioural alterations. Nat. Commun. 2014, 5, 4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Tan, Q.; Luo, Q. Decreased expression level and DNA-Binding activity of specificity protein 1 via cyclooxygenase-2 inhibition antagonizes radiation resistance, cell migration and invasion in radiation-resistant lung cancer cells. Oncol. Lett. 2018, 16, 3029–3037. [Google Scholar] [CrossRef]
- Yang, J.; Wei, D.; Liu, J. Repressions of MMP-9 expression and NF-κB localization are involved in inhibition of lung carcinoma 95-D cell invasion by (–)-epigallocatechin-3-gallate. Biomed. Pharmacother. 2005, 59, 98–103. [Google Scholar] [CrossRef]
- Nazir, S.U.; Kumar, R.; Singh, A.; Khan, A.; Tanwar, P.; Tripathi, R.; Mehrotra, R.; Hussain, S. Breast cancer invasion and progression by MMP-9 through Ets-1 transcription factor. Gene 2019, 711, 143952. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, L.D.; Sun, M.C.; Gu, W.D.; Geng, H.Z. Over-Expression of mir-124 inhibits MMP-9 expression and decreases invasion of renal cell carcinoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6308–6314. [Google Scholar] [CrossRef]
- Wang, W.; Yang, C.; Wang, X.Y.; Zhou, L.Y.; Lao, G.J.; Wang, C.; Liu, D.; Hu, M.D.; Zeng, T.T.; Yan, L.; et al. MicroRNA-129 and -335 Promote Diabetic Wound Healing by Inhibiting Sp1-Mediated MMP-9 Expression. Diabetes 2018, 67, 1627–1638. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Takino, T.; Endo, Y.; Sato, H. Activation of MMP-9 by membrane type-1 MMP/MMP-2 axis stimulates tumor metastasis. Cancer Sci. 2017, 108, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Gilardoni, M.B.; Remedi, M.M.; Oviedo, M.; DellaVedova, T.; Sarría, J.P.; Racca, L.; Dominguez, M.; Pellizas, C.G.; Donadio, A.C. Differential expression of Low density lipoprotein Receptor–Related Protein 1 (LRP-1) and matrix metalloproteinase-9 (MMP-9) in prostate gland: From normal to malignant lesions. Pathol. Res. Pract. 2017, 213, 66–71. [Google Scholar] [CrossRef]
- Chan, C.Y.; Chan, Y.C.; Cheuk, B.L.; Cheng, S.W. Clearance of matrix metalloproteinase-9 is dependent on low-Density lipoprotein receptor-Related protein-1 expression downregulated by microRNA-205 in human abdominal aortic aneurysm. J. Vasc. Surg. 2017, 65, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, R.; Ferrán, B.; Alonso, J.; Martí-Pàmies, Í.; Aguiló, S.; Calvayrac, O.; Rodríguez, C.; Martínez-González, J. NR4A receptors up-Regulate the antiproteinase alpha-2 macroglobulin (A2M) and modUlate MMP-2 and MMP-9 in vascular smooth muscle cells. Thromb. Haemost. 2015, 113, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, Q.; Wang, S.; Huang, Z.; Yu, F.; Lin, J. HER4 promotes the growth and metastasis of osteosarcoma via the PI3K/AKT pathway. Acta Biochim. Biophys. Sin. 2020, 52, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Pelekanou, V.; Villarroel-Espindola, F.; Schalper, K.A.; Pusztai, L.; Rimm, D.L. CD68, CD163, and matrix metalloproteinase 9 (MMP-9) co-localization in breast tumor microenvironment predicts survival differently in ER-positive and negative cancers. Breast Cancer Res. 2018, 20, 154. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Tang, F.; Zhang, B.; Zhao, Y.; Feng, J.; Rao, Z. Matrix metalloproteinase-9 overexpression is closely related to poor prognosis in patients with colon cancer. World J. Surg. Oncol. 2014, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.-Z.; Meng, X.-M.; Li, G.-S. Long non-coding RNA SNHG15 indicates poor prognosis of non-Small cell lung cancer and promotes cell proliferation and invasion. Eur. Rev. Med Pharmacol. Sci. 2018, 22, 2671–2679. [Google Scholar] [CrossRef]
- Li, Y.-M.; Liu, Z.-Y.; Li, Z.-C.; Wang, J.-C.; Yu, J.-M.; Yang, H.-J.; Chen, Z.-N.; Tang, J. Alterations of the Immunologic Co-Stimulator B7 and TNFR Families Correlate with Hepatocellular Carcinoma Prognosis and Metastasis by Inactivating STAT3. Int. J. Mol. Sci. 2019, 20, 156. [Google Scholar] [CrossRef] [Green Version]
- Barillari, G.; Monini, P.; Sgadari, C.; Ensoli, B. The Impact of Human Papilloma Viruses, Matrix Metallo-Proteinases and HIV Protease Inhibitors on the Onset and Progression of Uterine Cervix Epithelial Tumors: A Review of Preclinical and Clinical Studies. Int. J. Mol. Sci. 2018, 19, 1418. [Google Scholar] [CrossRef] [Green Version]
- Bedkowska, G.; Piskór, B.; Gacuta, E.; Zajkowska, M.; Osada, J.; Szmitkowski, M.; Dąbrowska, M.; Ławicki, S. Diagnostic Power of Selected Cytokines, MMPs and TIMPs in Ovarian Cancer Patients—ROC Analysis. Anticancer Res. 2019, 39, 2575–2582. [Google Scholar] [CrossRef]
- Pryczynicz, A.; Guzińska-Ustymowicz, K.; Dymicka-Piekarska, V.; Czyzewska, J.; Kemona, A. Expression of matrix metalloproteinase 9 in pancreatic ductal carcinoma is associated with tumor metastasis formation. Folia Histochem. Cytobiol. 2007, 45, 37–40. [Google Scholar]
- Yao, Z.; Yuan, T.; Wang, H.; Yao, S.; Zhao, Y.; Liu, Y.; Jin, S.; Chu, J.; Xu, Y.; Zhou, W.; et al. MMP-2 together with MMP-9 overexpression correlated with lymph node metastasis and poor prognosis in early gastric carcinoma. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghallab, N.A.; Shaker, O.G. Serum and salivary levels of chemerin and MMP-9 in oral squamous cell carcinoma and oral premalignant lesions. Clin. Oral Investig. 2016, 21, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-S.; Hung, T.-W.; Su, S.C.; Lin, C.-L.; Yang, S.-F.; Lee, C.-C.; Yeh, C.-F.; Hsieh, Y.-H.; Tsai, J.-P. MTA2 as a Potential Biomarker and Its Involvement in Metastatic Progression of Human Renal Cancer by miR-133b Targeting MMP-9. Cancers 2019, 11, 1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhang, S.; Chen, J. c-Myc induced upregulation of long non-coding RNA SNHG16 enhances progression and carcinogenesis in oral squamous cell carcinoma. Cancer Gene Ther. 2019, 26, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L.; et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition. Carcinogenesis 2015, 36, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype. Oxid. Med. Cell. Longev. 2018, 2018, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-Y.; Shen, J.-X.; Wang, Y.; Liu, Y.; Shen, D.-Y.; Quan, S. Tankyrase Promotes Aerobic Glycolysis and Proliferation of Ovarian Cancer through Activation of Wnt/β-Catenin Signaling. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Multhoff, G. Fatal Alliance of Hypoxia-/HIF-1α-Driven Microenvironmental Traits Promoting Cancer Progression. Adv. Exp. Med. Biol. 2020, 1232, 169–176. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Olea-Flores, M.O.; Zuñiga-Eulogio, M.D.; Mendoza-Catalan, M.A.; Rodríguez-Ruiz, H.A.; Castañeda-Saucedo, E.; Ortuno-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.-Y.; Hong, O.-Y.; Youn, H.J.; Kim, M.-G.; Kim, C.-H.; Jung, S.H. 15d-PGJ2 inhibits NF-κB and AP-1-mediated MMP-9 expression and invasion of breast cancer cell by means of a heme oxygenase-1-dependent mechanism. BMB Rep. 2020, 53, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Bove, P.F.; Wesley, U.V.; Greul, A.-K.; Hristova, M.; Dostmann, W.R.; Van Der Vliet, A. Nitric Oxide Promotes Airway Epithelial Wound Repair through Enhanced Activation of MMP-9. Am. J. Respir. Cell Mol. Biol. 2006, 36, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Jiang, J.; Huang, L.; Jiang, Y.; Yu, N.; Liu, X.; Lv, Y.; Li, H.; Zou, L.; Peng, C.; et al. iNOS is associated with tumorigenicity as an independent prognosticator in human intrahepatic cholangiocarcinoma. Cancer Manag. Res. 2019, 11, 8005–8022. [Google Scholar] [CrossRef] [Green Version]
- Sidibé, A.; Ropraz, P.; Jemelin, S.; Emre, Y.; Poittevin, M.; Pocard, M.; Bradfield, P.F.; Imhof, B.A. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat. Commun. 2018, 9, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2016, 17, 38–53. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Liapis, H.; Flath, A.; Kitazawa, S. Integrin alpha V beta 3 expression by bone-Residing breast cancer metastases. Diagn. Mol. Pathol. 1996, 5, 127–135. [Google Scholar] [CrossRef]
- Gasparini, G.; Brooks, P.C.; Biganzoli, E.; Vermeulen, P.B.; Bonoldi, E.; Dirix, L.Y.; Ranieri, G.; Miceli, R.; Cheresh, D.A. Vascular integrin alpha(v)beta3: A new prognostic indicator in breast cancer. Clin. Cancer Res. 1998, 4, 2625–2634. [Google Scholar]
- Weingarten, C.; Jenudi, Y.; Tshuva, R.Y.; Moskovich, D.; Alfandari, A.; Hercbergs, A.; Davis, P.J.; Ellis, M.; Ashur-Fabian, O. The Interplay between epithelial-Mesenchymal transition (EMT) and the thyroid hormones-αvβ3 axis in ovarian cancer. Horm. Cancer 2017, 9, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, A.M.; Becker, J.C.; Siu, C.H.; Lemmon, V.P.; Cheresh, D.A.; Pancook, J.D.; Zhao, X.; Reisfeld, R.A. Human neural cell adhesion molecule L1 and rat homologue NILE are ligands for integrin alpha v beta 3. J. Cell Biol. 1996, 132, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridiandries, A.; Tan, J.; Bursill, C.A. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1856. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Tolosano, E.; Munaron, L.; Genova, T. Targeting Metabolism to Counteract Tumor Angiogenesis: A Review of Patent Literature. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.H.; Gajjar, K.A.; Pearson, R.M.; Launiere, C.A.; Eddington, D.T.; Hong, S. Direct Measurements on CD24-Mediated Rolling of Human Breast Cancer MCF-7 Cells on E-Selectin. Anal. Chem. 2011, 83, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.N.; Zhu, F.; Schnaar, R.L.; Alves, C.S.; Konstantopoulos, K. Carcinoembryonic Antigen and CD44 Variant Isoforms Cooperate to Mediate Colon Carcinoma Cell Adhesion to E- and L-selectin in Shear Flow. J. Biol. Chem. 2008, 283, 15647–15655. [Google Scholar] [CrossRef] [Green Version]
- Sökeland, G.; Schumacher, U. The functional role of integrins during intra- and extravasation within the metastatic cascade. Mol. Cancer 2019, 18, 12. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genome Res. 1999, 13, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Liang, C.; Wang, S.; Zhu, J.; Miao, C.; Hua, Y.; Bao, M.; Cao, Q.; Qin, C.; Shao, P.; et al. CD151 promotes cell metastasis via activating TGF-β1/Smad signaling in renal cell carcinoma. Oncotarget 2018, 9, 13313–13323. [Google Scholar] [CrossRef]
- Hsu, C.C.; Huang, S.F.; Wang, J.S.; Chu, W.K.; Nien, J.E.; Chen, W.S.; Chow, SE. Interplay of N-cadherin and matrix metalloproteinase 9 enhances human nasopharyngeal carcinoma cell invasion. BMC Cancer 2016, 16, 800. [Google Scholar] [CrossRef] [Green Version]
- Criscuoli, M.L.; Nguyen, M.; Eliceiri, B.P. Tumor metastasis but not tumor growth is dependent on Src-mediated vascular permeability. Blood 2005, 105, 1508–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajska, A.; Jankowska-Steifer, E.; Czarnowska, E.; Olkowski, R.; Gula, G.; Niderla-Bielińska, J.; Flaht-Zabost, A.; Jasińska, A. Vasculogenesis and its cellular therapeutic applications. Cells Tissues Organs 2017, 203, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Tamma, R.; Ribatti, D. Bone Niches, Hematopoietic Stem Cells, and Vessel Formation. Int. J. Mol. Sci. 2017, 18, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiasson-MacKenzie, C.; McClatchey, A.I. Cell–Cell Contact and Receptor Tyrosine Kinase Signaling. Cold Spring Harb. Perspect. Biol. 2017, 10, a029215. [Google Scholar] [CrossRef]
- Yeung, K.T.; Yang, J. Epithelial-Mesenchymal transition in tumor metastasis. Mol. Oncol. 2016, 11, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-Mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, F.; Li, T.; Xiao, P.; Han, Z.-J.; Shu, L.-F.; Yuan, Z.-Z.; Liu, W.-J.; Long, Y.-Q. Extracellular Histone Promotes Prostate Cancer Migration and Epithelial-Mesenchymal Transition through NF-κB-Mediated Inflammatory Responses. Chemotherapy 2019, 64, 177–186. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E.; Aldo, P.; Elisabetta, C. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2019, 18, 343–351. [Google Scholar] [CrossRef]
- Kreiseder, B.; Orel, L.; Bujnow, C.; Buschek, S.; Pflueger, M.; Schuett, W.; Hundsberger, H.; Martin, R.; Wiesner, C. α-Catulin downregulates E-cadherin and promotes melanoma progression and invasion. Int. J. Cancer 2012, 132, 521–530. [Google Scholar] [CrossRef]
- Vinnakota, K.; Zhang, Y.; Selvanesan, B.C.; Topi, G.; Salim, T.; Sand-Dejmek, J.; Jönsson, G.; Sjölander, A. M2-like macrophages induce colon cancer cell invasion via matrix metalloproteinases. J. Cell. Physiol. 2017, 232, 3468–3480. [Google Scholar] [CrossRef]
- Vaisar, T.; Kassim, S.Y.; Gomez, I.G.; Green, P.S.; Hargarten, S.; Gough, P.J.; Parks, W.C.; Wilson, C.L.; Raines, E.W.; Heinecke, J.W. MMP-9 sheds the beta2 integrin subunit (CD18) from macrophages. Mol. Cell. Proteom. 2008, 8, 1044–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP generated matrikines. Matrix Biol. 2015, 44, 122–129. [Google Scholar] [CrossRef]
- Banyard, J.; Bielenberg, D.R. The role of EMT and MET in cancer dissemination. Connect. Tissue Res. 2015, 56, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, S.-L.; Hsieh, S.; Lai, P.-Y.; Wang, J.-J.; Li, C.-C.; Wu, C.-C. Carnosine Suppresses Human Colorectal Cell Migration and Intravasation by Regulating EMT and MMP Expression. Am. J. Chin. Med. 2019, 47, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, M.R.; Varricchi, G.; Loffredo, S.; Bellevicine, C.; Lansione, T.; Ferrara, A.L.; Iannone, R.; Di Somma, S.; Borriello, F.; Clery, E.; et al. Potential involvement of neutrophils in human thyroid cancer. PLoS ONE 2018, 13, e0199740. [Google Scholar] [CrossRef]
- Wu, L.; Awaji, M.; Saxena, S.; Varney, M.L.; Sharma, B.; Singh, R.K. IL-17-CXC Chemokine Receptor 2 Axis Facilitates Breast Cancer Progression by Up-Regulating Neutrophil Recruitment. Am. J. Pathol. 2019, 190, 222–233. [Google Scholar] [CrossRef]
- Reymond, N.; d’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal "Liaisons" during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells—Mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2016, 14, 155–167. [Google Scholar] [CrossRef]
- Shiraishi, K.; Mimura, K.; Kua, L.-F.; Koh, V.; Siang, L.K.; Nakajima, S.; Fujii, H.; Shabbir, A.; Yong, W.-P.; So, J.B.; et al. Inhibition of MMP activity can restore NKG2D ligand expression in gastric cancer, leading to improved NK cell susceptibility. J. Gastroenterol. 2016, 51, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Henschen-Edman, A.H.; Nagase, H.; Tenner, A.J. Digestion of C1q collagen-Like domain with MMPs-1, -2,- 3, and -9 further defines the sequence involved in the stimulation of neutrophil superoxide production. J. Leukoc. Biol. 1999, 66, 416–422. [Google Scholar] [CrossRef]
- Dabagh, M.; Gounley, J.; Randles, A. Localization of Rolling and Firm-Adhesive Interactions Between Circulating Tumor Cells and the Microvasculature Wall. Cell. Mol. Bioeng. 2020, 13, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Voura, E.B.; English, J.L.; Yu, H.-Y.; Van Ho, A.T.; Subarsky, P.; Hill, R.P.; Hojilla, C.V.; Khokha, R. Proteolysis during Tumor Cell Extravasation In Vitro: Metalloproteinase Involvement across Tumor Cell Types. PLoS ONE 2013, 8, e78413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.V.; Gutierrez, L.M.; Correa, A.; Lazarowski, A.; Bolontrade, M. Metastatic Niches and the Modulatory Contribution of Mesenchymal Stem Cells and Its Exosomes. Int. J. Mol. Sci. 2019, 20, 1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Shi, H.; Gao, Y.; Qiao, X.; Li, H.; Wei, M.; Zeng, X. Lung resided monocytic myeloid-derived suppressor cells contribute to premetastatic niche formation by enhancing MMP-9 expression. Mol. Cell. Probes 2020, 50, 101498. [Google Scholar] [CrossRef]
- Vered, M.; Shnaiderman-Shapiro, A.; Schiby, G.; Zlotogorski-Hurvitz, A.; Salo, T.; Yahalom, R. Markers of the pre-Metastatic niche “knock on the door” of metastasis-Free cervical lymph nodes in patients with oral cancer. Acta Histochem. 2019, 121, 151447. [Google Scholar] [CrossRef]
- Wu, S.; Zheng, Q.; Xing, X.; Dong, Y.; Wang, Y.; You, Y.; Chen, R.; Hu, C.; Chen, J.; Gao, D.; et al. Matrix stiffness-Upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-Metastatic niche formation. J. Exp. Clin. Cancer Res. 2018, 37, 99. [Google Scholar] [CrossRef]
- Li, M.; Lu, Y.; Xu, Y.; Wang, J.; Zhang, C.; Du, Y.; Wang, L.; Li, L.; Wang, B.; Shen, J.; et al. Horizontal transfer of exosomal CXCR4 promotes murine hepatocarcinoma cell migration, invasion and lymphangiogenesis. Gene 2018, 676, 101–109. [Google Scholar] [CrossRef]
- Li, Y.Y.; Tao, Y.W.; Gao, S.; Li, P.; Zheng, J.M.; Zhang, S.E.; Liang, J.; Zhang, Y. Cancer-Associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-Mediated paracrine miR-34a-5p. EBioMedicine 2018, 36, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Yang, F.; Yu, L.; Yu, Z.; Jiang, L.; Wang, Q.; Yang, Y.; Wang, L.; Cao, X.; Wang, J. Activated T Cell Exosomes Promote Tumor Invasion via Fas Signaling Pathway. J. Immunol. 2012, 188, 5954–5961. [Google Scholar] [CrossRef] [Green Version]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2004, 113, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, H.; Xu, C.; Zhang, Y.; Zhang, Q.; Chen, L.; Ding, Q.; Deng, Z. Exosomal miR-106b serves as a novel marker for lung cancer and promotes cancer metastasis via targeting PTEN. Life Sci. 2020, 244, 117297. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Cui, X.; Lu, L.; Chen, G.; Yang, Y.; Hu, Y.; Lu, Y.; Cao, Z.; Wang, Y.; Wang, X. Exosomes from glioma cells induce a tumor-Like phenotype in mesenchymal stem cells by activating glycolysis. Stem Cell Res. Ther. 2019, 10, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linton, S.S.; Abraham, T.; Liao, J.; Clawson, G.A.; Butler, P.J.; Fox, T.; Kester, M.; Matters, G.L. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-Derived macrophages. PLoS ONE 2018, 13, e0206759. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z.; et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2018, 37, 242. [Google Scholar] [CrossRef] [Green Version]
- Mutschelknaus, L.; Azimzadeh, O.; Heider, T.; Winkler, K.; Vetter, M.; Kell, R.; Tapio, S.; Merl-Pham, J.; Huber, S.M.; Edalat, L.; et al. Radiation alters the cargo of exosomes released from squamous head and neck cancer cells to promote migration of recipient cells. Sci. Rep. 2017, 7, 12423. [Google Scholar] [CrossRef]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.E.; Sanderson, R.D. Fibronectin on the Surface of Myeloma Cell-Derived Exosomes Mediates Exosome-Cell Interactions. J. Biol. Chem. 2015, 291, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Qin, H.; Poon, T.C.; Sze, S.-C.; Ding, X.; Na Co, N.; Ngai, S.-M.; Chan, T.-F.; Wong, N. Hepatocellular carcinoma-Derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015, 36, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Zhang, Y.; Wu, X. 786-0 Renal cancer cell line-Derived exosomes promote 786-0 cell migration and invasion in vitro. Oncol. Lett. 2014, 7, 1576–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell. Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Luo, L.; Bai, X.; Shen, K.; Liu, K.; Wang, J.; Hu, D. Highly-Expressed micoRNA-21 in adipose derived stem cell exosomes can enhance the migration and proliferation of the HaCaT cells by increasing the MMP-9 expression through the PI3K/AKT pathway. Arch. Biochem. Biophys. 2020, 681, 108259. [Google Scholar] [CrossRef] [PubMed]
- Runz, S.; Keller, S.; Rupp, C.; Stoeck, A.; Issa, Y.; Koensgen, D.; Mustea, A.; Sehouli, J.; Kristiansen, G.; Altevogt, P. Malignant ascites-Derived exosomes of ovarian carcinoma patients contain CD24 and EpCAM. Gynecol. Oncol. 2007, 107, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Joshi, N.; Choi, H.; Ryu, S.; Hahn, M.; Catena, R.; Sadik, H.; Argani, P.; Wagner, P.; Vahdat, L.; et al. Myeloid Progenitor Cells in the Premetastatic Lung Promote Metastases by Inducing Mesenchymal to Epithelial Transition. Cancer Res. 2012, 72, 1384–1394. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-Mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-Mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.K.; Kim, M.J.; Jang, H.S.; Lee, H.R.; Ahn, K.M.; Lee, J.H.; Choung, P.H.; Kim, M.J. A high concentration of MMP-2/gelatinase A and MMP-9/gelatinase B reduce NK cell-Mediated cytotoxicity against an oral squamous cell carcinoma cell line. In Vivo 2008, 22, 593–597. [Google Scholar]
- Peng, Y.-P.; Zhang, J.-J.; Liang, W.-B.; Tu, M.; Lu, Z.; Wei, J.; Jiang, K.; Gao, W.; Wu, J.; Xu, Z.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, V.; O’Sullivan, C.; Stefanutti, E.; Kovalenko, M.; Greenstein, A.; Barry-Hamilton, V.; Mikaelian, I.; Degenhardt, J.; Yue, P.; Smith, V.; et al. MMP-9 inhibition promotes anti-Tumor immunity through disruption of biochemical and physical barriers to T-Cell trafficking to tumors. PLoS ONE 2018, 13, e0207255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkan, D.; Green, J.E.; Chambers, A. Extracellular matrix: A gatekeeper in the transition from dormancy to metastatic growth. Eur. J. Cancer 2010, 46, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, H.; Yan, L.; Du, W.; Zhang, M.; Chen, H.; Zhang, L.; Li, G.; Li, J.; Dong, Y.; et al. MMP-2 and MMP-9 contribute to the angiogenic effect produced by hypoxia/15-HETE in pulmonary endothelial cells. J. Mol. Cell. Cardiol. 2018, 121, 36–50. [Google Scholar] [CrossRef]
- Naik, M.U.; Naik, U.M. Junctional adhesion molecule-A-Induced endothelial cell migration on vitronectin is integrin alpha v beta 3 specific. J. Cell Sci. 2006, 119, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-J.; Yang, Y.; Yang, G.-K.; Wan, J.; Cui, D.-L.; Ma, Z.-H.; Du, L.-J.; Zhang, G.-M. Slit2 suppresses endothelial cell proliferation and migration by inhibiting the VEGF-Notch signaling pathway. Mol. Med. Rep. 2017, 15, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.A.; Kang, M.H.; Lee, H.J.; Kim, B.-H.; Park, J.K.; Kim, H.K.; Kim, J.S.; Oh, S.C. Sonic Hedgehog Pathway Promotes Metastasis and Lymphangiogenesis via Activation of Akt, EMT, and MMP-9 Pathway in Gastric Cancer. Cancer Res. 2011, 71, 7061–7070. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.T. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc. Res. 2006, 69, 677–687. [Google Scholar] [CrossRef]
- Hirte, H.; Vergote, I.; Jeffrey, J.; Grimshaw, R.; Coppieters, S.; Schwartz, B.; Tu, D.; Sadura, A.; Brundage, M.; Seymour, L. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: A National Cancer Institute of Canada Clinical Trials Group Study. Gynecol. Oncol. 2006, 102, 300–308. [Google Scholar] [CrossRef]
- Tan, B.L.; Esa, N.M. Curcumin Combination Chemotherapy: The Implication and Efficacy in Cancer. MolEcules 2019, 24, 2527. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.A.; Starodub, A.N.; Sharma, S.; Berlin, J.; Patel, M.R.; Wainberg, Z.A.; Chaves, J.; Gordon, M.S.; Windsor, K.; Brachmann, C.B.; et al. Andecaliximab/GS-5745 Alone and Combined with mFOLFOX6 in Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: Results from a Phase I Study. Clin. Cancer Res. 2018, 24, 3829–3837. [Google Scholar] [CrossRef] [Green Version]
- Addison, C.; Simos, D.; Wang, Z.; Pond, G.; Smith, S.; Robertson, S.; Mazzarello, S.; Singh, G.; VanderMeer, L.; Fernandes, R.; et al. A phase 2 trial exploring the clinical and correlative effects of combining doxycycline with bone-targeted therapy in patients with metastatic breast cancer. J. Bone Oncol. 2016, 5, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Hampson, L.; Maranga, I.O.; Masinde, M.S.; Oliver, A.W.; Batman, G.; He, X.; Desai, M.; Okemwa, P.M.; Stringfellow, H.; Martin-Hirsch, P.; et al. A Single-Arm, Proof-of-Concept Trial of Lopimune (Lopinavir/Ritonavir) as a Treatment for HPV-Related Pre-Invasive Cervical Disease. PLoS ONE 2016, 11, e0147917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastos, K.; Lu, D.; Shi, Q.; Tien, P.C.; Kaplan, R.C.; Hessol, N.A.; Cole, S.; Vigen, C.; Cohen, M.; Young, M.; et al. Association of Serum Lipid Levels With HIV Serostatus, Specific Antiretroviral Agents, and Treatment Regimens. J. Acquir. Immune Defic. Syndr. 2007, 45, 34–42. [Google Scholar] [CrossRef]
- Mulligan, K.; Grunfeld, C.; Tai, V.W.; Algren, H.; Pang, M.; Chernoff, D.N.; Lo, J.C.; Schambelan, M. Hyperlipidemia and Insulin Resistance Are Induced by Protease Inhibitors Independent of Changes in Body Composition in Patients With HIV Infection. J. Acquir. Immune Defic. Syndr. 2000, 23, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kchour, G.; Tarhini, M.; Kooshyar, M.M.; El Hajj, H.; Wattel, E.; Mahmoudi, M.; Hatoum, H.; Rahimi, H.; Maleki, M.; Rafatpanah, H.; et al. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-Cell leukemia/lymphoma (ATL). Blood 2009, 113, 6528–6532. [Google Scholar] [CrossRef] [PubMed]
- Leroi, C.; Balestre, E.; Messou, E.; Minga, A.; Sawadogo, A.; Drabo, J.; Maïga, M.; Zannou, M.; Seydi, M.; Dabis, F.; et al. Incidence of Severe Neutropenia in HIV-Infected People Starting Antiretroviral Therapy in West Africa. PLoS ONE 2017, 12, e0170753. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, P.K.; Fiala, M.A.; Grossman, B.J.; DiPersio, J.F.; Stockerl-Goldstein, K.; Gao, F.; Uy, G.L.; Westervelt, P.; Schroeder, M.A.; Cashen, A.F.; et al. Results of a Prospective Randomized, Open-Label, Noninferiority Study of Tbo-Filgrastim (Granix) versus Filgrastim (Neupogen) in Combination with Plerixafor for Autologous Stem Cell Mobilization in Patients with Multiple Myeloma and Non-Hodgkin Lymphoma. Biol. Blood Marrow Transpl. 2017, 23, 2065–2069. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Cuadrón, D.; Boluda, B.; Martinez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I-II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef]
- Cashen, A.F.; Rettig, M.; Gao, F.; Smith, A.; Abboud, C.; Stockerl-Goldstein, K.; Vij, R.; Uy, G.L.; Westervelt, P.; DiPersio, J. Phase I/II Study of Intravenous Plerixafor Added to a Mobilization Regimen of Granulocyte Colony–Stimulating Factor in Lymphoma Patients Undergoing Autologous Stem Cell Collection. Biol. Blood Marrow Transpl. 2017, 23, 1282–1289. [Google Scholar] [CrossRef]
- Neuwirthová, J.; Gal, B.; Smilek, P.; Urbánková, P. Potential of the Flavonoid Quercetin to Prevent and Treat Cancer–Current Status of Research. Klin. Onkol. 2018, 31, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisele, G.; Wick, A.; Eisele, A.-C.; Clement, P.M.; Tonn, J.; Tabatabai, G.; Ochsenbein, A.; Schlegel, U.; Neyns, B.; Krex, D.; et al. Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression. J. Neurooncol. 2014, 117, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mira, E.; LaCalle, R.A.; Buesa, J.M.; De Buitrago, G.G.; Jiménez-Baranda, S.; Gómez-Moutón, C.; Martínez-A, C.; Manes, S. Secreted MMP9 promotes angiogenesis more efficiently than constitutive active MMP9 bound to the tumor cell surface. J. Cell Sci. 2004, 117, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wang, X.; Zhao, Y.; Curtis, J.M.; Brindley, D.N. Doxycycline attenuates breast cancer related inflammation by decreasing plasma lysophosphatidate concentrations and inhibiting NF-κB activation. Mol. Cancer 2017, 16, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górska, R.; Nedzi-Gora, M. The effects of the initial treatment phase and of adjunctive low-dose doxycycline therapy on clinical parameters and MMP-8, MMP-9, and TIMP-1 levels in the saliva and peripheral blood of patients with chronic periodontitis. Arch. Immunol. Ther. Exp. 2006, 54, 419–426. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2017, 175, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Monini, P.; Sgadari, C.; Toschi, E.; Barillari, G.; Ensoli, B. Antitumour effects of antiretroviral therapy. Nat. Rev. Cancer 2004, 4, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Beaulieu, R.; Gelmon, K.; Thuot, C.A.; Sawka, C.; Read, S.; Singer, J. Prospective randomized trial of two dose levels of interferon alfa with zidovudine for the treatment of Kaposi’s sarcoma associated with human immunodeficiency virus infection: A Canadian HIV Clinical Trials Network study. J. Clin. Oncol. 1998, 16, 1736–1742. [Google Scholar] [CrossRef]
- Monini, P.; Sgadari, C.; Grosso, M.G.; Bellino, S.; Di Biagio, A.; Toschi, E.; Bacigalupo, I.; Sabbatucci, M.; Cencioni, G.; Salvi, E.; et al. Clinical course of classic Kaposi’s sarcoma in HIV-Negative patients treated with the HIV protease inhibitor indinavir. AIDS 2009, 23, 534–538. [Google Scholar] [CrossRef]
- Houédé, N.; Pulido, M.; Mourey, L.; Joly, F.; Ferrero, J.-M.; Bellera, C.A.; Priou, F.; Lalet, C.; Laroche-Clary, A.; Raffin, M.C.; et al. A Phase II Trial Evaluating the Efficacy and Safety of Efavirenz in Metastatic Castration-Resistant Prostate Cancer. Oncologist 2014, 19, 1227–1228. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Hann, H.-W.; Coben, R.; Conn, M.; Dimarino, A.J. Update Treatment for HBV Infection and Persistent Risk for Hepatocellular Carcinoma: Prospect for an HBV Cure. Diseases 2018, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Toschi, E.; Sgadari, C.; Malavasi, L.; Bacigalupo, I.; Chiozzini, C.; Carlei, D.; Compagnoni, D.; Bellino, S.; Bugarini, R.; Falchi, M.; et al. Human immunodeficiency virus protease inhibitors reduce the growth of human tumors via a proteasome-Independent block of angiogenesis and matrix metalloproteinases. Int. J. Cancer 2010, 128, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Esposito, V.; Verdina, A.; Manente, L.; Spugnini, E.P.; Viglietti, R.; Parrella, R.; Pagliano, P.; Parrella, G.; Galati, R.; De Luca, A.; et al. Amprenavir inhibits the migration in human hepatocarcinoma cell and the growth of xenografts. J. Cell. Physiol. 2012, 228, 640–645. [Google Scholar] [CrossRef]
- Song, S.; Ji, Y.; Zhang, G.; Zhang, X.; Li, B.; Li, D.; Jiang, W. Protective Effect of Atazanavir Sulphate Against Pulmonary FibrosisIn VivoandIn Vitro. Basic Clin. Pharmacol. Toxicol. 2017, 122, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, K.T.; Lin, C.-L.; Cella, M.; McMichael, A.J.; Austyn, J.M.; Rowland-Jones, S.L. The HIV protease inhibitor Indinavir reduces immature dendritic cell transendothelial migration. Eur. J. Immunol. 2003, 33, 2520–2530. [Google Scholar] [CrossRef]
- Sanchez, C.G.; Molinski, S.; Gongora, R.; Sosulski, M.; Fuselier, T.; MacKinnon, S.S.; Mondal, D.; Lasky, J.A. The antiretroviral agent nelfinavir mesylate: a potential therapy for systemic sclerosis. Arthritis Rheumatol. 2017, 70, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hresko, R.C.; Hruz, P.W. HIV Protease Inhibitors Act as Competitive Inhibitors of the Cytoplasmic Glucose Binding Site of GLUTs with Differing Affinities for GLUT1 and GLUT4. PLoS ONE 2011, 6, e25237. [Google Scholar] [CrossRef] [Green Version]
- Richmond, S.R.; Carper, M.J.; Lei, X.; Zhang, S.; Yarasheski, K.; Ramanadham, S. HIV-Protease inhibitors suppress skeletal muscle fatty acid oxidation by reducing CD36 and CPT1 fatty acid transporters. Biochim. Biophys. Acta 2010, 1801, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacigalupo, I.; Palladino, C.; Leone, P.; Toschi, E.; Sgadari, C.; Ensoli, B.; Barillari, G. Inhibition of MMP-9 expression by ritonavir or saquinavir is associated with inactivation of the AKT/Fra-1 pathway in cervical intraepithelial neoplasia cells. Oncol. Lett. 2017, 13, 2903–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Barros, S.; Zakaroff-Girard, A.; Lafontan, M.; Galitzky, J.; Bourlier, V. Inhibition of human preadipocyte proteasomal activity by HIV protease inhibitors or specific inhibitor lactacystin leads to a defect in adipoGenesis, which involves MMP-9. J. Pharmacol. Exp. Ther. 2006, 320, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Latronico, T.; Liuzzi, G.M.; Riccio, P.; Lichtner, M.; Mengoni, F.; D’Agostino, C.; Vullo, V.; Mastroianni, C.M. Antiretroviral therapy inhibits matrix metalloproteinase-9 from blood mononuclear cells of HIV-infected patients. AIDS 2007, 21, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Hennessy, M.; Bergin, C.; Mulcahy, F.; Hopkins, S.; Spiers, J.P. Ribavirin and interferon alter MMP-9 abundance in vitro and in HIV-HCV-Coinfected patients. Antivir. Ther. 2011, 16, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latronico, T.; Pati, I.; Ciavarella, R.; Fasano, A.; Mengoni, F.; Lichtner, M.; Vullo, V.; Mastroianni, C.M.; Liuzzi, G.M. In vitro effect of antiretroviral drugs on cultured primary astrocytes: Analysis of neurotoxicity and matrix metalloproteinase inhibition. J. Neurochem. 2018, 144, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liuzzi, G.M.; Mastroianni, C.M.; Latronico, T.; Mengoni, F.; Fasano, A.; Lichtner, M.; Vullo, V.; Riccio, P. Anti-HIV drugs decrease the expression of matrix metalloproteinases in astrocytes and microglia. Brain 2004, 127, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV Protease Inhibitors Decrease VEGF/HIF-1α Expression and Angiogenesis in Glioblastoma Cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir down-Regulates hypoxia-Inducible factor 1alpha and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef] [Green Version]
- Ikezoe, T.; Saito, T.; Bandobashi, K.; Yang, Y.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-Regulated kinase 1/2. Mol. Cancer Ther. 2004, 3, 473–479. [Google Scholar]
- Hong-Brown, L.Q.; Brown, C.R.; Lang, C.H. Indinavir impairs protein synthesis and phosphorylations of MAPKs in mouse C2C12 myocytes. Am. J. Physiol. Physiol. 2004, 287, C1482–C1492. [Google Scholar] [CrossRef] [Green Version]
- Ravaud, C.; Paré, M.; Yao, X.; Azoulay, S.; Mazure, N.M.; Dani, C.; Ladoux, A. Resveratrol and HIV-protease inhibitors control UCP1 expression through opposite effects on p38 MAPK phosphorylation in human adipocytes. J. Cell. Physiol. 2019, 235, 1184–1196. [Google Scholar] [CrossRef]
- Wallet, M.A.; Reist, C.M.; Williams, J.C.; Appelberg, S.; Guiulfo, G.L.; Gardner, B.; Sleasman, J.W.; Goodenow, M.M. The HIV-1 protease inhibitor nelfinavir activates PP2 and inhibits MAPK signaling in macrophages: A pathway to reduce inflammation. J. Leukoc. Biol. 2012, 92, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.; Bikas, A.; Patel, A.; Kushchayeva, Y.; Costello, J.; McDaniel, D.; Burman, K.; Vasko, V. Nelfinavir inhibits proliferation and induces DNA damage in thyroid cancer cells. Endocr. Relat. Cancer 2017, 24, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; See, A.P.; Aziz, K.; Thiyagarajan, S.; Salih, T.; Gajula, R.P.; Armour, M.; Phallen, J.; Terezakis, S.; Kleinberg, L.; et al. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biol. Ther. 2011, 12, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Bryant, C.S.; Chamala, S.; Qazi, A.; Seward, S.M.; Pal, J.; Steffes, C.P.; Weaver, D.W.; Morris, R.; Malone, J.M.; et al. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol. Cancer 2009, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Lee, J.H.; Wilke, W.W.; Quon, H.; Smith, G.; Maity, A.; Buatti, J.M.; Spitz, D.R. Radiation Response in Two HPV-Infected Head-and-Neck Cancer Cell Lines in Comparison to a Non–HPV-Infected Cell Line and Relationship to Signaling Through AKT. Int. J. Radiat. Oncol. 2009, 74, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Pore, N.; Cerniglia, G.J.; Mick, R.; Georgescu, M.-M.; Bernhard, E.J.; Hahn, S.; Gupta, A.K.; Maity, A. Phosphatase and Tensin Homologue Deficiency in Glioblastoma Confers Resistance to Radiation and Temozolomide that Is Reversed by the Protease Inhibitor Nelfinavir. Cancer Res. 2007, 67, 4467–4473. [Google Scholar] [CrossRef] [Green Version]
- Srirangam, A.; Mitra, R.; Wang, M.; Gorski, J.C.; Badve, S.S.; Baldridge, L.A.; Hamilton, J.; Kishimoto, H.; Hawes, J.; Li, L.; et al. Effects of HIV protease inhibitor ritonavir on Akt-Regulated cell proliferation in breast cancer. Clin. Cancer Res. 2006, 12, 1883–1896. [Google Scholar] [CrossRef] [Green Version]
- Batchu, R.B.; Gruzdyn, O.V.; Bryant, C.S.; Qazi, A.; Kumar, S.; Chamala, S.; Kung, S.T.; Sanka, R.S.; Puttagunta, U.S.; Weaver, D.W.; et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals 2014, 7, 46–57. [Google Scholar] [CrossRef]
- Pasquereau, S.; Kumar, A.; Abbas, W.; Herbein, G. Counteracting Akt Activation by HIV Protease Inhibitors in Monocytes/Macrophages. Viruses 2018, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Plastaras, J.P.; Vapiwala, N.; Ahmed, M.S.; Gudonis, D.; Cerniglia, G.J.; Feldman, M.D.; Frank, I.; Gupta, A.K. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 2008, 7, 628–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Romano, R.; Rudich, A.; Tirosh, A.; Potashnik, R.; Sasaoka, T.; Riesenberg, K.; Schlaeffer, F.; Bashan, N. Nelfinavir-Induced insulin resistance is associated with impaired plasma membrane recruitment of the PI3-kinase effectors AKT/PKB and PKC-Zeta. Diabetologia 2004, 47, 1107–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachko, I.; Maissel, A.; Mazor, L.; Ben-Romano, R.; Watson, R.T.; Hou, J.C.; Pessin, J.E.; Bashan, N.; Rudich, A. Postreceptoral adipocyte insulin resistance induced by nelfinavir is caused by insensitivity of PKB/Akt to phosphatidylinositol-3,4,5-trisphosphate. Endocrinology 2009, 150, 2618–2626. [Google Scholar] [CrossRef] [Green Version]
- Barillari, G.; Iovane, A.; Bacigalupo, I.; Labbaye, C.; Chiozzini, C.; Sernicola, L.; Quaranta, M.T.; Falchi, M.; Sgadari, C.; Ensoli, B. The HIV protease inhibitor indinavir down-Regulates the expression of the pro-angiogenic MT1-MMP by human endothelial cells. Angiogenesis 2014, 17, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mukherji, M.; Mandal, A. Ritonavir inhibits HIF-1α-Mediated VEGF expression in retinal pigment epithelial cells in vitro. Eye 2013, 28, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Equils, O.; Shapiro, A.; Madak-Erdogan, Z.; Liu, C.; Lu, D. Human Immunodeficiency Virus Type 1 Protease Inhibitors Block Toll-Like Receptor 2 (TLR2)- and TLR4-Induced NF-κB Activation. Antimicrob. Agents Chemother. 2004, 48, 3905–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Zhang, X.; Huang, H.; Ji, Y.; Li, D.; Jiang, W. Saquinavir plus methylprednisolone ameliorates experimental acute lung injury. Braz. J. Med. Biol. Res. 2018, 51, 7579. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Zhang, G.; Zhu, H.; Li, D.; Jiang, W. Indinavir Plus Methylprednisolone Ameliorates Experimental Acute Lung Injury In Vitro and In Vivo. Shock 2018, 49, 196–204. [Google Scholar] [CrossRef]
- Malizia, A.P.; Cotter, E.; Chew, N.; Powderly, W.G.; Doran, P. HIV Protease Inhibitors Selectively Induce Gene Expression Alterations Associated with Reduced Calcium Deposition in Primary Human Osteoblasts. AIDS Res. Hum. Retrovir. 2007, 23, 243–250. [Google Scholar] [CrossRef]
- Auclair, M.; Afonso, P.; Capel, E.; Caron-Debarle, M.; Capeau, J. Impact of darunavir, atazanavir and lopinavir boosted with ritonavir on cultured human endothelial cells: Beneficial effect of pravastatin. Antivir. Ther. 2014, 19, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.-M.; Jiang, J.; Jamaluddin, S.; Liang, Z.; Yao, Q.; Chen, C. Ginsenoside Rb1 Blocks Ritonavir-Induced Oxidative Stress and eNOS Downregulation through Activation of Estrogen Receptor-Beta and Upregulation of SOD in Human Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 294. [Google Scholar] [CrossRef] [Green Version]
- Chai, H.; Yang, H.; Yan, S.; Li, M.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Effects of 5 HIV Protease Inhibitors on Vasomotor Function and Superoxide Anion Production in Porcine Coronary Arteries. JAIDS J. Acquir. Immune Defic. Syndr. 2005, 40, 12–19. [Google Scholar] [CrossRef]
- Gary-Bobo, G.; Houssaini, A.; Amsellem, V.; Rideau, D.; Pacaud, P.; Perrin, A.; Brégeon, J.; Marcos, E.; Dubois-Randé, J.-L.; Sitbon, O.; et al. Effects of HIV protease inhibitors on progression of monocrotaline- and hypoxia-induced pulmonary hypertension in rats. Circulation 2010, 122, 1937–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-J.; Lin, S.-H.; Din, Z.-H.; Su, J.-H.; Liu, C.-I. Sinulariolide Inhibits Gastric Cancer Cell Migration and Invasion through Downregulation of the EMT Process and Suppression of FAK/PI3K/AKT/mTOR and MAPKs Signaling Pathways. Mar. Drugs 2019, 17, 668. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Wan, D.; Song, W. Dryofragin inhibits the migration and invasion of human osteosarcoma U2OS cells by suppressing MMP-2/9 and elevating TIMP-1/2 through PI3K/AKT and p38 MAPK signaling pathways. Anticancer Drugs 2016, 27, 1–668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ma, J.; Mao, Y.; Dong, L.; Li, S.; Zhang, Y. Silence of α1-Antitrypsin Inhibits Migration and Proliferation of Triple Negative Breast Cancer Cells. Med. Sci. Monit. 2018, 24, 6851–6860. [Google Scholar] [CrossRef]
- Canani, R.B.; Spagnuolo, M.I.; Cirillo, P.; Guarino, A. Ritonavir Combination Therapy Restores Intestinal Function in Children With Advanced HIV Disease. J. Acquir. Immune Defic. Syndr. 1999, 21, 307–312. [Google Scholar] [CrossRef]
- Sgadari, C.; Barillari, G.; Toschi, E.; Carlei, D.; Bacigalupo, I.; Baccarini, S.; Palladino, C.; Leone, P.; Bugarini, R.; Malavasi, L.; et al. HIV protease inhibitors are potent anti-Angiogenic molecules and promote regression of Kaposi sarcoma. Nat. Med. 2002, 8, 225–232. [Google Scholar] [CrossRef]
- Loizzi, V.; Del Vecchio, V.; Gargano, G.; De Liso, M.; Kardashi, A.; Naglieri, E.; Resta, L.; Cicinelli, E.; Cormio, G. Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. Int. J. Mol. Sci. 2017, 18, 1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbab, A.S. Activation of alternative pathways of angiogenesis and involvement of stem cells following anti-angiogenesis treatment in glioma. Histol. Histopathol. 2012, 27, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hill, E.J.; Roberts, C.; Franklin, J.M.; Enescu, M.; West, N.; MacGregor, T.P.; Chu, K.-Y.; Boyle, L.; Blesing, C.; Wang, L.M.; et al. Clinical Trial of Oral Nelfinavir before and during Radiation Therapy for Advanced Rectal Cancer. Clin. Cancer Res. 2016, 22, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.; Fokas, E.; Dutton, S.J.; Patel, N.; Hawkins, M.A.; Eccles, C.; Chu, K.-Y.; Durrant, L.; Abraham, A.G.; Partridge, M.; et al. ARCII: A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother. Oncol. 2016, 119, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conant, M.A.; Opp, K.M.; Poretz, D.; Mills, R.G. Reduction of Kaposi’s sarcoma lesions following treatment of AIDS with ritonovir. AIDS 1997, 11, 1300–1301. [Google Scholar] [CrossRef] [PubMed]
- Lebbé, C.; Blum, L.; Pellet, C.; Blanchard, G.; Verola, O.; Morel, P.; Danne, O.; Calvo, F. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-Related Kaposi’s sarcoma. AIDS 1998, 12, F45–F49. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Patton, C.; Stevens, G.; Tekautz, T.; Angelov, L.; Vogelbaum, M.A.; Weil, R.J.; Chao, S.; Elson, P.; Suh, J.H.; et al. Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-Grade gliomas. J. Neurooncol. 2010, 102, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.C.; Milhem, M.; Anderson, C.; Sun, W.; Smith, B.J.; Hoffman, H.T.; Buatti, J.M. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: Results of a phase II clinical trial. Head Neck 2014, 37, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yu, H.; Sun, F.-Y.; Li, J.; Li, D.-M.; Sun, C.-H. Effects of lamivudine on cell proliferation of liver cancer and expressions of HBsAg, HBeAg, and MMP-9 in patients. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9093–9098. [Google Scholar] [PubMed]
- Song, L.; Ding, S.; Ge, Z.; Zhu, X.; Qiu, C.; Wang, Y.; Lai, E.; Yang, W.; Sun, Y.; Chow, S.A.; et al. Nucleoside/nucleotide reverse transcriptase inhibitors attenuate angiogenesis and lymphangiogenesis by impairing receptor tyrosine kinases signalling in endothelial cells. Br. J. Pharmacol. 2017, 175, 1241–1259. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chai, H.; Lin, P.H.; Yao, Q.; Chen, C. Roles and Mechanisms of Human Immunodeficiency Virus Protease Inhibitor Ritonavir and Other Anti-Human Immunodeficiency Virus Drugs in Endothelial Dysfunction of Porcine Pulmonary Arteries and Human Pulmonary Artery Endothelial Cells. Am. J. Pathol. 2009, 174, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Jamaluddin, S.; Lin, P.H.; Yao, Q.; Chen, C. Non-Nucleoside reverse transcriptase inhibitor efavirenz increases monolayer permeability of human coronary artery endothelial cells. Atherosclerosis 2009, 208, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Matłosz, B.; Kowalska, J.D.; Bąkowska, E.; Firląg-Burkacka, E.F.-B.; Vassilenko, A.; Horban, A. Discontinuation of tenofovir due to nephrotoxicity: Insight into 12 years of clinical practice. Przegl. Epidemiol. 2019, 73, 249–255. [Google Scholar] [CrossRef]
- Quercia, R.; Perno, C.-F.; Koteff, J.; Moore, K.; McCoig, C.; Clair, M.S.; Kuritzkes, D. Twenty-Five Years of Lamivudine. J. Acquir. Immune Defic. Syndr. 2018, 78, 125–135. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Mozobil (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir. Chem. Chemother. 2019, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinni, S.R.; Sivalogan, S.; Dong, Z.; Filho, J.C.T.; Deng, X.; Bonfil, R.D.; Cher, M.L. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: The role of bone microenvironment-Associated CXCL12. Prostate 2006, 66, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, A.M.; Seoane, S.; Cabezas, P.; Eiro, N.; Lago, J.J.S.; Macia, M.; García-Caballero, T.; Gonzalez, L.O.; Sánchez, L.; Vizoso, F.; et al. Breast cancer metastasis to liver and lung is facilitated by Pit-1-CXCL12-CXCR4 axis. Oncogene 2018, 37, 1430–1444. [Google Scholar] [CrossRef]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Y.; Zhang, S.; Chen, Z.; Fan, L.; Shen, X.; Zhou, S.; Chen, D. Stem cell autocrine CXCL12/CXCR4 stimulates invasion and metastasis of esophageal cancer. Oncotarget 2017, 8, 36149–36160. [Google Scholar] [CrossRef] [Green Version]
- Mao, T.L.; Fan, K.F.; Liu, C.L. Targeting the CXCR4/CXCL12 axis in treating epithelial ovarian cancer. Gene Ther. 2017, 24, 621–629. [Google Scholar] [CrossRef]
- Stanisavljević, L.; Aßmus, J.; Storli, K.E.; Leh, S.; Dahl, O.; Myklebust, M.P. CXCR4, CXCL12 and the relative CXCL12-CXCR4 expression as prognostic factors in colon cancer. Tumor Biol. 2015, 37, 7441–7452. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Muzi, P.; Ventura, L.; Biordi, L.; Ricevuto, E.; Pompili, S.; Mattei, C.; Di Cesare, E.; Jannini, E.A.; et al. CXCR4 pharmacogical inhibition reduces bone and soft tissue metastatic burden by affecting tumor growth and tumorigenic potential in prostate cancer preclinical models. Prostate 2015, 75, 1227–1246. [Google Scholar] [CrossRef]
- Werner, T.A.; Forster, C.M.; Dizdar, L.; Verde, P.E.; Raba, K.; Schott, M.; Knoefel, W.T.; Krieg, A. CXCR4/CXCR7/CXCL12-Axis in Follicular Thyroid Carcinoma. J. Cancer 2018, 9, 929–940. [Google Scholar] [CrossRef]
- Chaudary, N.; Pintilie, M.; Jelveh, S.; Lindsay, P.; Hill, R.P.; Milosevic, M. Plerixafor Improves Primary Tumor Response and Reduces Metastases in Cervical Cancer Treated with Radio-Chemotherapy. Clin. Cancer Res. 2016, 23, 1242–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, A.; Hsu, S.H.; Blanco, A.; Buryanek, J.; Day, A.L.; McGuire, M.F.; Brown, R.E. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience 2016, 3, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Driessen, C.; Müller, R.; Novak, U.; Cantoni, N.; Betticher, D.; Mach, N.; Rüfer, A.; Mey, U.; Samaras, P.; Ribi, K.; et al. Promising activity of nelfinavir-Bortezomib-Dexamethasone in proteasome inhibitor–refractory multiple myeloma. Blood 2018, 132, 2097–2100. [Google Scholar] [CrossRef]
- De Weger, V.A.; Stuurman, F.E.; Hendrikx, J.J.; Moes, J.J.; Sawicki, E.; Huitema, A.D.R.; Nuijen, B.; Thijssen, B.; Rosing, H.; Keessen, M.; et al. A dose-Escalation study of bi-Daily once weekly oral docetaxel either as ModraDoc001 or ModraDoc006 combined with ritonavir. Eur. J. Cancer 2017, 86, 217–225. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]



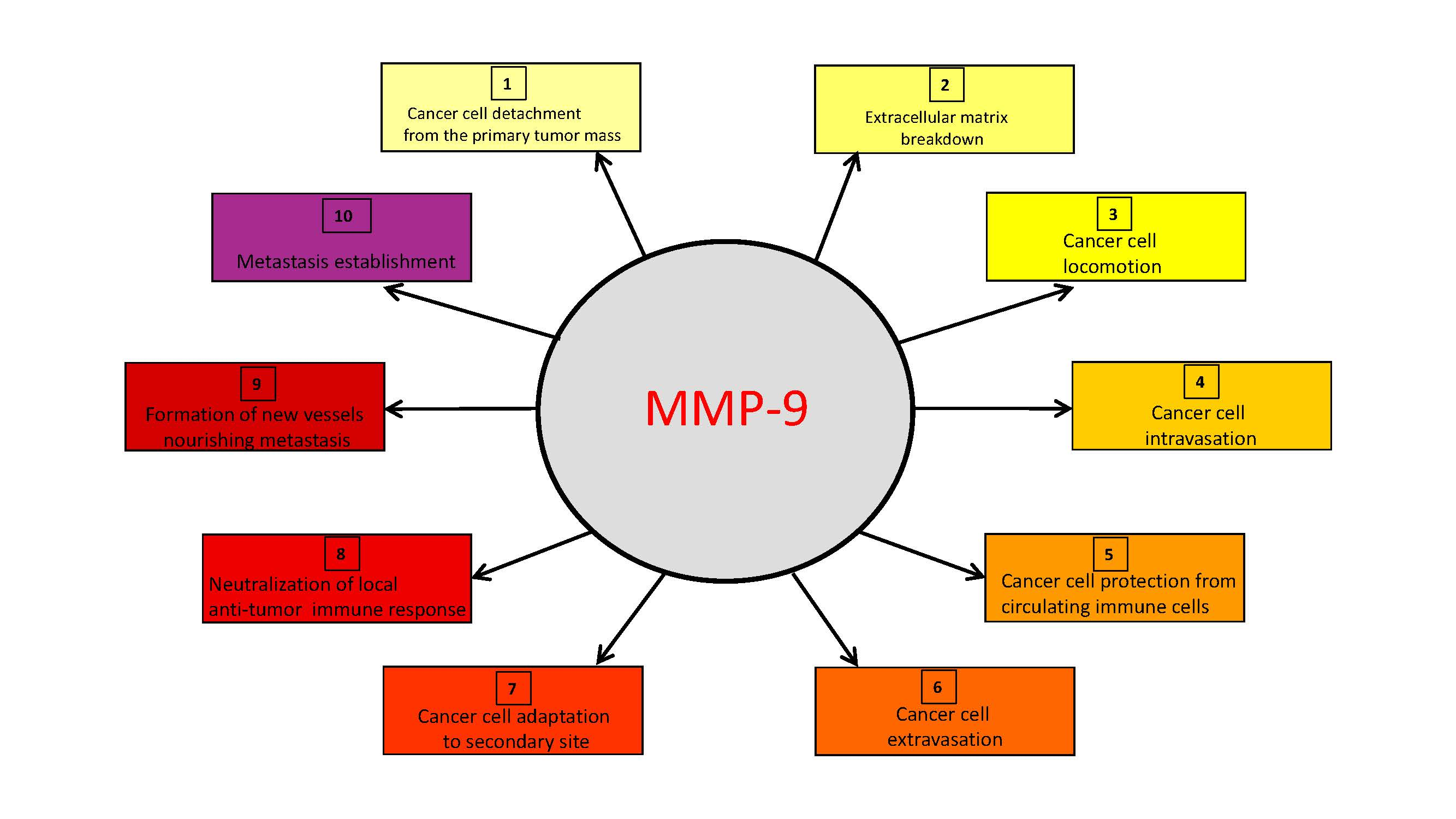{kind=link}
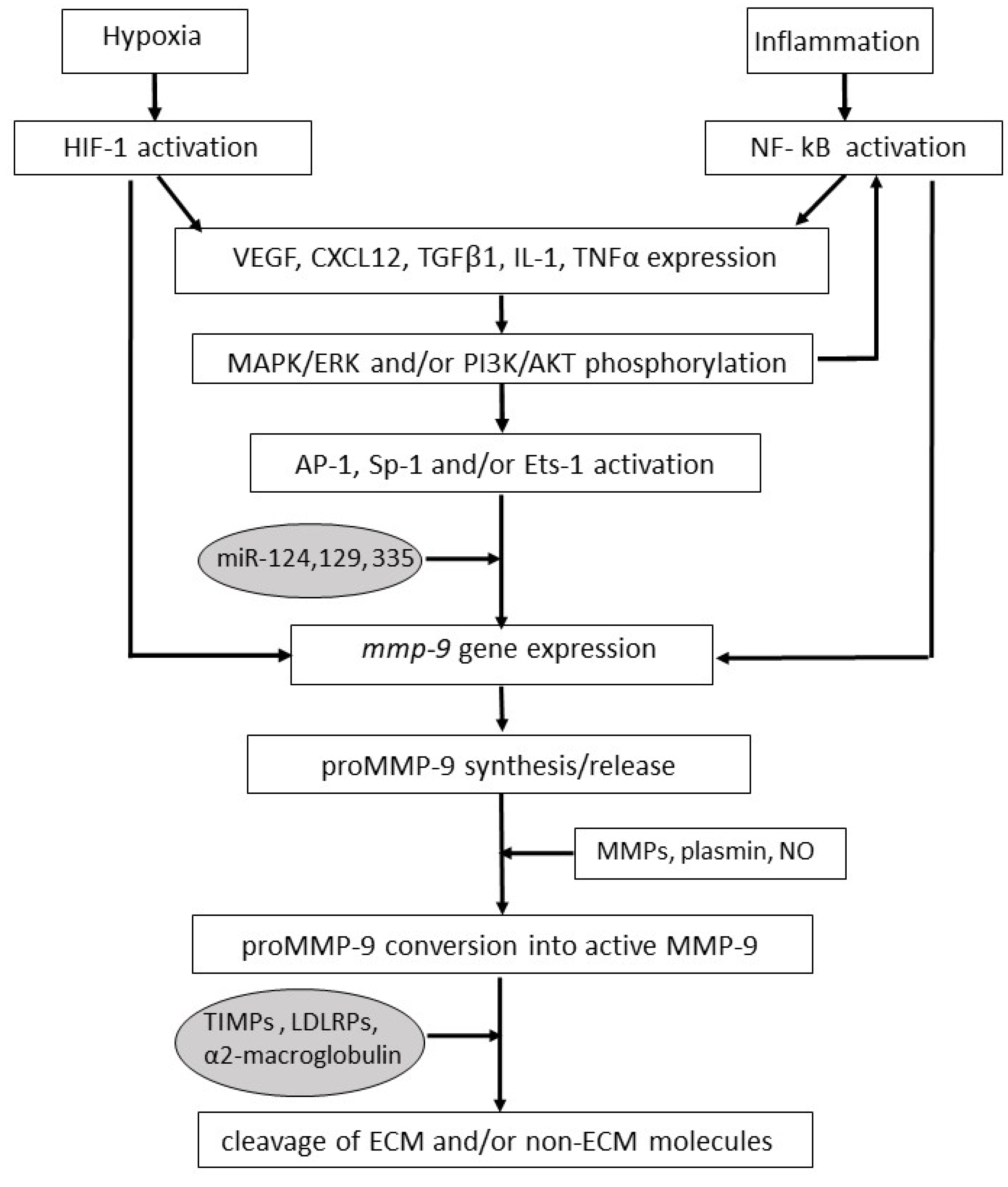{kind=link}
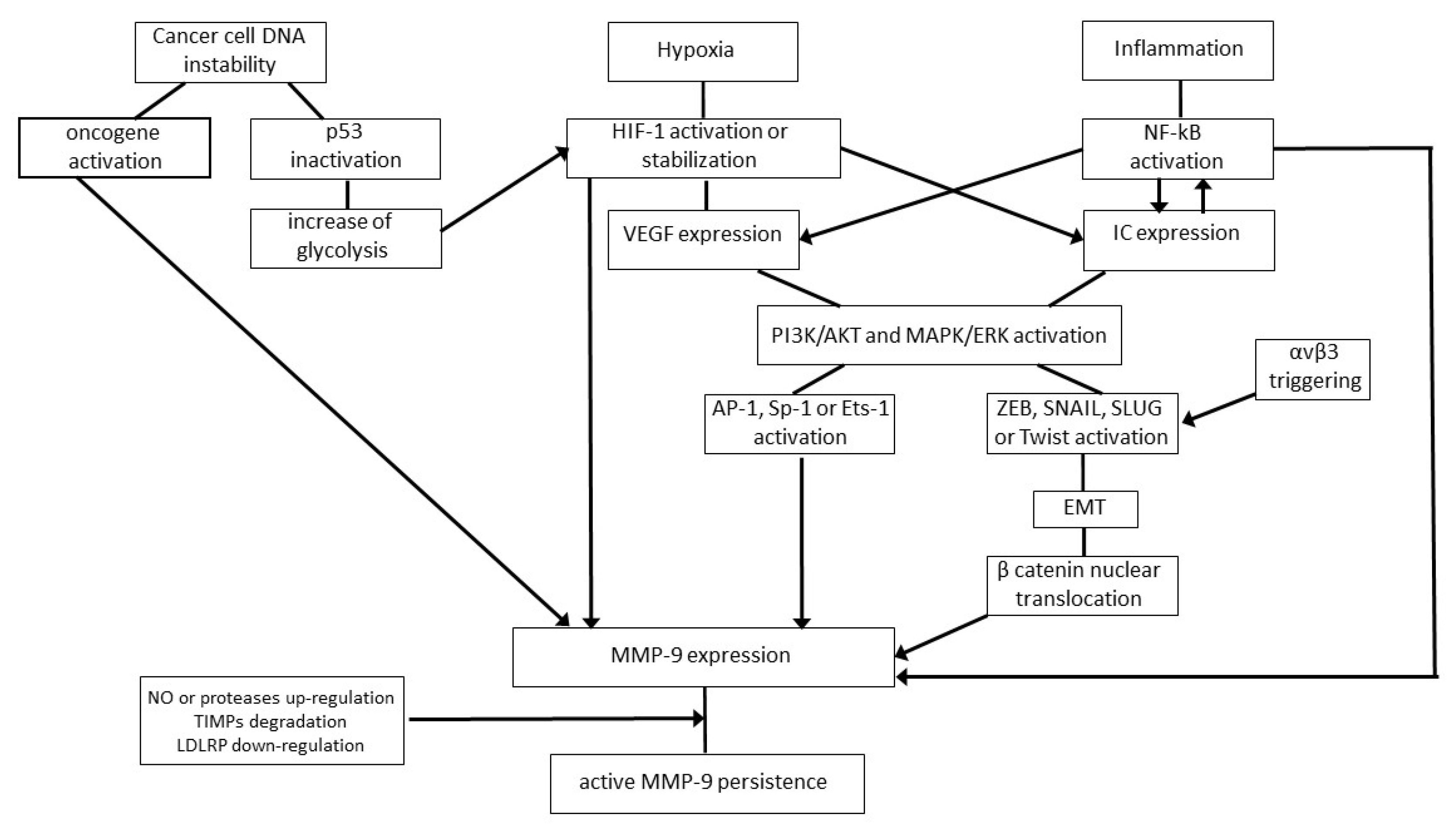{kind=link}
| Molecule | Action |
| Nitric oxide | Contributes to proMMP-9 conversion into active MMP-9 [63,64] |
| Tryspin or chymotrypsin | Degrade TIMPs [67] |
| MMP-2 and MT1-MMP | Activate cell surface-bound MMP-9 [39] |
| Plasmin or MMP-1, 3, 7, 10, 13 and 26 | Activate released MMP-9 [4] |
| αvβ3 | Drives tumor or endothelial cell migration toward chemotactic factors generated and/or released by MMP-9 [68,69,70,71]; Facilitates MMP-9-promoted cancer cell extravasion [72]; Cooperates with MMP-9 to induce new vessel formation [73,74] |
| α4β1, CEA or CD24 | Facilitate MMP-9-promoted cancer cell extravasion [75,76,77]. |
| CD44 | Facilitates MMP-9-promoted cancer cell extravasion and spatially directs MMP-9 proteolytic activity [78]. |
| CD151 | Facilitates MMP-9-promoted cancer cell locomotion [79]. |
| Neuronal-cadherin | Facilitates the dissemination of cancer cells promoted by MMP-9 [80] |
| VEGF | Cooperates with MMP-9 to induce cancer cell transendothelial migration [81], and new vessel formation [73,74,82] |
| CXCL12 | Cooperates with MMP-9 in promoting new vessel formation [73,74,83] |
| Drug | Class | Mechanism of Action | Clinical Outcomes | Toxicities |
|---|---|---|---|---|
| MARIMASTAT | Broad-spectrum MMPI | Chelates MMP catalytic zinc | Does not improve OS of glioblastoma, NSCLC and pancreatic or colorectal ca pts [138] | Musculoskeletal pain [138] |
| TANOMASTAT | Narrow-spectrum MMPI | Binds MMP exosite | Does not improve OS of NSCLC, pancreatic or ovarian ca pts [139] | Nausea, vomiting [139] |
| CURCUMIN | Plant extract | Reduces MMP-9 expression | Improves the efficacy of chemotherapy in prostate ca pts [140] | None [140] |
| ANDECALIXIMAB | Humanized mAB | Neutralizes MMP-9 | Improves OS and reduces tumor size in gastric ca pts [141] | Nausea, vomiting, fatigue [141] |
| DOXYCYCLINE | Antibiotic | Reduces MMP-9 expression | Does not improve OS of breast ca pts [142] | Nausea, vomiting, diarrhea [142] |
| LPV-RTV | HIV-PI | Inhibits the AKT-MMP-9 pathway | effective against CIN [143] | Increase in glycaemia and/or lipemia [144,145] |
| AZT | HIV-RTI | Reduces MMP-9 expression | Improves the efficacy of chemotherapy in T cell leukemia pts [146] | Headache, nausea, vomiting, neutropenia, anemia, hepatotoxicity, myopathy [147] |
| AMD3100 | CXCR4 antagonist | Inhibits the CXCL12-AKT-MMP-9 pathway | Effective against hematological malignancies [148,149] | Musculoskeletal pain, gastrointestinal disturbances [150] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barillari, G. The Impact of Matrix Metalloproteinase-9 on the Sequential Steps of the Metastatic Process. Int. J. Mol. Sci. 2020, 21, 4526. https://doi.org/10.3390/ijms21124526
Barillari G. The Impact of Matrix Metalloproteinase-9 on the Sequential Steps of the Metastatic Process. International Journal of Molecular Sciences. 2020; 21(12):4526. https://doi.org/10.3390/ijms21124526
Chicago/Turabian StyleBarillari, Giovanni. 2020. "The Impact of Matrix Metalloproteinase-9 on the Sequential Steps of the Metastatic Process" International Journal of Molecular Sciences 21, no. 12: 4526. https://doi.org/10.3390/ijms21124526
APA StyleBarillari, G. (2020). The Impact of Matrix Metalloproteinase-9 on the Sequential Steps of the Metastatic Process. International Journal of Molecular Sciences, 21(12), 4526. https://doi.org/10.3390/ijms21124526