An Overview of In Vivo and In Vitro Models for Autosomal Dominant Polycystic Kidney Disease: A Journey from 3D-Cysts to Mini-Pigs

 ,
,  and
and
Abstract
:1. Introduction
- suitability regarding the specific research hypothesis under study,
- relevance to the human pathology in terms of causative mechanisms and symptoms,
- availability and ease of manipulation (e.g., tools already available and well established in the laboratory conducting the research vs. introduction of new technologies),
- economic factors and rapidness of result obtention.
2. In Vitro Research Tools
2.1. Cultured Monolayers of Kidney Cells and Three-Dimensional Cysts
2.2. Embryonic Kidney Culture
2.3. Stem Cell Approaches and Kidney Organoids
3. In Vivo Research Models
3.1. Invertebrates
3.2. Lower Vertebrates
3.3. Drug Screening Approaches in Invertebrate and Lower Vertebrate Models of ADPKD
3.4. Rodent Models
3.5. Other Mammalian Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2/3D | 2/3-dimensional |
| 2DG | 2Deoxy-d-glucose |
| ADPKD | Autosomal dominant polycystic kidney disease |
| ARPKD | Autosomal recessive polycystic kidney disease |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| ECM | Extracellular matrix |
| ESC | Embryonic stem cells |
| ESRD | End-stage renal disease |
| Gnas | Guanine nucleotide binding protein G subunit α isoform (short) |
| hESC | Human embryonic stem cell |
| hPSC | Human pluripotent stem cell |
| iPSC | Induced pluripotent stem cell |
| ISMR | International Mouse Strain Resource |
| MDCK | Madin–Darby canine kidney |
| PC-1/2 | Polycystin 1/2 |
| PKD | Polycystic kidney disease |
| PPE1A | Phosphodiesterase 1A |
References
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Grantham, J.J. Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2008, 359, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, S.; Blanchette, C.; Claxton, A.; Roy, D.; Rossetti, S.; Gutierrez, B. End-stage renal disease in autosomal dominant polycystic kidney disease: A comparison of dialysis-related utilization and costs with other chronic kidney diseases. Clin. Outcomes Res. 2015, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Grantham, J.J.; Torres, V.E.; Chapman, A.B.; Guay-Woodford, L.M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Baumgarten, D.A.; Kenney, P.J.; Harris, P.C.; et al. Volume Progression in Polycystic Kidney Disease. N. Engl. J. Med. 2006, 354, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- van den Dool, S.W.; Wasser, M.N.; de Fijter, J.W.; Hoekstra, J.; van der Geest, R.J. Functional Renal Volume: Quantitative Analysis at Gadolinium-enhanced MR Angiography—Feasibility Study in Healthy Potential Kidney Donors. Radiology 2005, 236, 189–195. [Google Scholar] [CrossRef]
- Spithoven, E.M.; Kramer, A.; Meijer, E.; Orskov, B.; Wanner, C.; Caskey, F.; Collart, F.; Finne, P.; Fogarty, D.G.; Groothoff, J.W.; et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014, 86, 1244–1252. [Google Scholar] [CrossRef] [Green Version]
- Luciano, R.L.; Dahl, N.K. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): Considerations for routine screening and management. Nephrol. Dial. Transpl. 2014, 29, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Cai, Q.; Guo, X.-Y.; Bai, D.-H.; Sheng, H.-Z.; Wang, B.-K.; Yan, K.; Lu, A.-M.; Wang, X.-R. Effectiveness of Tolvaptan in the Treatment for Patients with Autosomal Dominant Polycystic Kidney Disease: A Meta-analysis. Comb. Chem. High Throughput Screen 2020, 23, 6–16. [Google Scholar] [CrossRef]
- Blair, H.A. Tolvaptan: A Review in Autosomal Dominant Polycystic Kidney Disease. Drugs 2019, 79, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sans-Atxer, L.; Joly, D. Tolvaptan in the treatment of autosomal dominant polycystic kidney disease: Patient selection and special considerations. Int. J. Nephrol. Renov. Dis. 2018, 11, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowley, B.D., Jr. Polycystic Kidney Disease Progression: Learning from Europe. Am. J. Nephrol. 2018, 48, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Ward, C.J.; Peral, B.; Aspinwall, R.; Clark, K.; San Millán, J.L.; Gamble, V.; Harris, P.C. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 1995, 10, 151–160. [Google Scholar] [CrossRef]
- The International Polycystic Kidney Disease Consortium. Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell 1995, 81, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.-P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Ong, A.C.M.; Harris, P.C. A polycystin-centric view of cyst formation and disease: The polycystins revisited. Kidney Int. 2015, 88, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Saigusa, T.; Bell, P.D. Molecular Pathways and Therapies in Autosomal-Dominant Polycystic Kidney Disease. Physiology 2015, 30, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Harris, P.C. Mechanisms of Disease: Autosomal dominant and recessive polycystic kidney diseases. Nat. Clin. Pr. Nephrol. 2006, 2, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Mangos, S.; Lam, P.; Zhao, A.; Liu, Y.; Mudumana, S.; Vasilyev, A.; Liu, A.; Drummond, I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model. Mech. 2010, 3, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekahli, D.; Parys, J.B.; Bultynck, G.; Missiaen, L.; De Smedt, H. Polycystins and cellular Ca2+ signaling. Cell. Mol. Life Sci. 2013, 70, 2697–2712. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.M.; Vanden Heuvel, G.B. Kidney: Polycystic kidney disease. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 465–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvadori, M.; Tsalouchos, A. Novel Therapeutic Strategies Targeting Molecular Pathways of Cystogenesis in Autosomal Polycystic Kidney Disease. J. Ren. Hepatic Disord. 2017, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Menezes, L.F.; Germino, G.G. The pathobiology of polycystic kidney disease from a metabolic viewpoint. Nat. Rev. Nephrol. 2019, 15, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Eccles, M.R.; Stayner, C.A. Polycystic kidney diseas—Where gene dosage counts. F1000Prime Rep. 2014, 6, 1–6. [Google Scholar] [CrossRef]
- Happé, H.; Peters, D.J.M. Translational research in ADPKD: Lessons from animal models. Nat. Rev. Nephrol. 2014, 10, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Takakura, A.; Contrino, L.; Zhou, X.; Bonventre, J.V.; Sun, Y.; Humphreys, B.D.; Zhou, J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum. Mol. Genet. 2009, 18, 2523–2531. [Google Scholar] [CrossRef] [Green Version]
- Weimbs, T. Third-hit signaling in renal cyst formation. J. Am. Soc. Nephrol. 2011, 22, 793–795. [Google Scholar] [CrossRef]
- Kurbegovic, A.; Trudel, M. Acute kidney injury induces hallmarks of polycystic kidney disease. Am. J. Physiol. Physiol. 2016, 311, F740–F751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, N.M.; Freedman, B.S. “Kidney in a Dish” Organoids for PKD. In Polycystic Kidney Disease; Hu, J., Yu, Y., Eds.; CRC Press: New York, NY, USA, 2019; pp. 177–194. ISBN 9780429468834. [Google Scholar]
- Weydert, C.; Decuypere, J.P.; De Smedt, H.; Janssens, P.; Vennekens, R.; Mekahli, D. Fundamental insights into autosomal dominant polycystic kidney disease from human-based cell models. Pediatr. Nephrol. 2019, 34, 1697–1715. [Google Scholar] [CrossRef] [PubMed]
- DesRochers, T.M.; Palma, E.; Kaplan, D.L. Tissue-engineered kidney disease models. Adv. Drug Deliv. Rev. 2014, 69–70, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e4. [Google Scholar] [CrossRef] [Green Version]
- Boreström, C.; Jonebring, A.; Guo, J.; Palmgren, H.; Cederblad, L.; Forslöw, A.; Svensson, A.; Söderberg, M.; Reznichenko, A.; Nyström, J.; et al. A CRISP(e)R view on kidney organoids allows generation of an induced pluripotent stem cell-derived kidney model for drug discovery. Kidney Int. 2018, 94, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling physiological events in 2D vs. 3D cell culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Dixon, E.E.; Woodward, O.M. Three-dimensional in vitro models answer the right questions in ADPKD cystogenesis. Am. J. Physiol. Physiol. 2018, 315, F332–F335. [Google Scholar] [CrossRef]
- Giuliani, S.; Perin, L.; Sedrakyan, S.; Kokorowski, P.; Jin, D.; De Filippo, R. Ex Vivo Whole Embryonic Kidney Culture: A Novel Method for Research in Development, Regeneration and Transplantation. J. Urol. 2008, 179, 365–370. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, H.; Yang, B. Drug discovery for polycystic kidney disease. Acta Pharm. Sin. 2011, 32, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Stulberg, C.S.; Coriell, L.L.; Kniazeff, A.J.; Shannon, J.E. The animal cell culture collection. In Vitro 1970, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- ATCC. MDCK (NBL-2) ATCC® CCL-34TM Canis Familiaris Kidney Normal. Available online: https://www.lgcstandards-atcc.org/products/all/CCL-34.aspx#history (accessed on 7 November 2019).
- Taub, M.; Chuman, L.; Saier, M.H.; Sato, G. Growth of Madin-Darby canine kidney epithelial cell (MDCK) line in hormone-supplemented, serum-free medium. Proc. Natl. Acad. Sci. USA 1979, 76, 3338–3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAteer, J.A.; Evan, A.P.; Gardner, K.D. Morphogenetic clonal growth of kidney epithelial cell line MDCK. Anat. Rec. 1987, 217, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J.; Uchic, M.; Cragoe, E.J.; Kornhaus, J.; Grantham, J.A.; Donoso, V.; Mangoo-Karim, R.; Evan, A.; McAteer, J. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int. 1989, 35, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Reif, G.A.; Wallace, D.P. Chapter 5: In vitro cyst formation of ADPKD cells. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2019; Volume 153, pp. 93–111. ISBN 9780128170823. [Google Scholar]
- Mangoo-Karim, R.; Uchic, M.; Grant, M.; Shumate, W.A.; Calvet, J.P.; Park, C.H.; Granthamt, J.J. Renal epithelial fluid secretion and cyst growth: The role of cyclic AMP. FASEB J. 1989, 3, 2629–2632. [Google Scholar] [CrossRef]
- Grantham, J.J.; Geiser, J.L.; Evan, A.P. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987, 31, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Mangoo-Karim, R.; Uchic, M.; Lechene, C.; Grantham, J.J. Renal epithelial cyst formation and enlargement in vitro: Dependence on cAMP. Proc. Natl. Acad. Sci. USA 1989, 86, 6007–6011. [Google Scholar] [CrossRef] [Green Version]
- Grantham, J.J.; Ye, M.; Gattone, V.H.; Sullivan, L.P. In vitro fluid secretion by epithelium from polycystic kidneys. J. Clin. Invest. 1995, 95, 195–202. [Google Scholar] [CrossRef]
- Davidow, C.J.; Maser, R.L.; Rome, L.A.; Calvet, J.P.; Grantham, J.J. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996, 50, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Lebeau, C.; Hanaoka, K.; Moore-Hoon, M.L.; Guggino, W.B.; Beauwens, R.; Devuyst, O. Basolateral chloride transporters in autosomal dominant polycystic kidney disease. Pflug. Arch. Eur. J. Physiol. 2002, 444, 722–731. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D.; Pelling, J.C.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belibi, F.A.; Reif, G.; Wallace, D.P.; Yamaguchi, T.; Olsen, L.; Li, H.; Helmkamp, G.M.; Grantham, J.J. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells11See Editorial by Torres. Kidney Int. 2004, 66, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reif, G.A.; Yamaguchi, T.; Nivens, E.; Fujiki, H.; Pinto, C.S.; Wallace, D.P. Tolvaptan inhibits ERK-dependent cell proliferation, Cl− secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am. J. Physiol. Ren. Physiol. 2011, 301, F1005–F1013. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.M.; Jacobs, D.T.; Allard, B.A.; Fields, T.A.; Sharma, M.; Wallace, D.P.; Tran, P. V Inhibition of Hedgehog signaling suppresses proliferation and microcyst formation of human Autosomal Dominant Polycystic Kidney Disease cells. Sci. Rep. 2018, 8, 4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idowu, J.; Home, T.; Patel, N.; Magenheimer, B.; Tran, P.V.; Maser, R.L.; Ward, C.J.; Calvet, J.P.; Wallace, D.P.; Sharma, M. Aberrant Regulation of Notch3 Signaling Pathway in Polycystic Kidney Disease. Sci. Rep. 2018, 8, 3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoo-Karim, R.; Grantham, J.J. Transepithelial water permeability in an in vitro model of renal cysts. J. Am. Soc. Nephrol. 1990, 1, 278–285. [Google Scholar]
- Wilson, P.D.; Sherwood, A.C.; Palla, K.; Du, J.; Watson, R.; Norman, J.T. Reversed polarity of Na (+) −K (+) -ATPase: Mislocation to apical plasma membranes in polycystic kidney disease epithelia. Am. J. Physiol. Physiol. 1991, 260, F420–F430. [Google Scholar] [CrossRef]
- Grant, M.E.; Neufeld, T.K.; Cragoe, E.J.; Welling, L.W.; Granthamt, J.J. Arginine Vasopressin Stimulates Net Fluid Secretion in a Polarized Subculture of Cyst-Forming MDCK Cells. J. Am. Soc. Nephrol. 1991, 2, 219–227. [Google Scholar]
- Neufeld, T.K.; Grant, M.E.; Grantham, J.J. A method to measure the rate of net fluid secretion by monolayers of cultured renal epithelial cells. J. Tissue Cult. Methods 1991, 13, 229–234. [Google Scholar] [CrossRef]
- Neufeld, T.K.; Douglass, D.; Grant, M.; Ye, M.; Silva, F.; Nadasdy, T.; Grantham, J.J. In vitro formation and expansion of cysts derived from human renal cortex epithelial cells. Kidney Int. 1992, 41, 1222–1236. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Grant, M.; Sharma, M.; Elzinga, L.; Swan, S.; Torres, V.E.; Grantham, J.J. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J. Am. Soc. Nephrol. 1992, 3, 984–994. [Google Scholar] [PubMed]
- Ye, M.; Grantham, J.J. The secretion of fluid by renal cysts from patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 1993, 329, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Booij, T.H.; Bange, H.; Leonhard, W.N.; Yan, K.; Fokkelman, M.; Kunnen, S.J.; Dauwerse, J.G.; Qin, Y.; van de Water, B.; van Westen, G.J.P.; et al. High-Throughput Phenotypic Screening of Kinase Inhibitors to Identify Drug Targets for Polycystic Kidney Disease. SLAS Discov. Adv. Life Sci. RD 2017, 22, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felekkis, K.N.; Koupepidou, P.; Kastanos, E.; Witzgall, R.; Bai, C.X.; Li, L.; Tsiokas, L.; Gretz, N.; Deltas, C. Mutant polycystin-2 induces proliferation in primary rat tubular epithelial cells in a STAT-1/p21-independent fashion accompanied instead by alterations in expression of p57KIP2 and Cdk2. BMC Nephrol. 2008, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asawa, R.R.; Danchik, C.; Zahkarov, A.; Chen, Y.; Voss, T.; Jadhav, A.; Wallace, D.P.; Trott, J.F.; Weiss, R.H.; Simeonov, A.; et al. A high-throughput screening platform for Polycystic Kidney Disease (PKD) drug repurposing utilizing murine and human ADPKD cells. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Avner, E.D.; Ellis, D.; Temple, T.; Jaffe, R. Metanephric development in serum-free organ culture. In Vitro 1982, 18, 675–682. [Google Scholar] [CrossRef]
- Yang, B.; Sonawane, N.D.; Zhao, D.; Somlo, S.; Verkman, A.S. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Magenheimer, B.S.; St John, P.L.; Isom, K.S.; Abrahamson, D.R.; De Lisle, R.C.; Wallace, D.P.; Maser, R.L.; Grantham, J.J.; Calvet, J.P. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+), K(+),2Cl(−) Co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 2006, 17, 3424–3437. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, F.; Sun, Y.; Lei, L.; Zhou, H.; Lei, T.; Xia, Y.; Verkman, A.S.; Yang, B. Aquaporin-1 retards renal cyst development in polycystic kidney disease by inhibition of Wnt signaling. FASEB J. 2015, 29, 1551–1563. [Google Scholar] [CrossRef] [Green Version]
- De Souza, N. Organoids. Nat. Methods 2018, 15, 23. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Becroft, M.; Vanslambrouck, J.M.; Stanley, E.G.; Elefanty, A.G.; Little, M.H. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 2014, 16, 118–126. [Google Scholar] [CrossRef]
- Morizane, R.; Bonventre, J. V Kidney Organoids: A Translational Journey. Trends Mol. Med. 2017, 23, 246–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Nam, S.A.; Yang, C.W. Applications of kidney organoids derived from human pluripotent stem cells. Korean J. Intern. Med. 2018, 33, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Thatava, T.; Armstrong, A.S.; De Lamo, J.; Edukulla, R.; Khan, Y.; Sakuma, T.; Ohmine, S.; Sundsbak, J.L.; Harris, P.C.; Kudva, Y.C.; et al. Successful disease-specific induced pluripotent stem cell generation from patients with kidney transplantation. Stem Cell Res. 2011, 2, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman Lab–Lab. Website of Benjamin, S.; Freedman, Ph.D. Available online: https://freedmanlab.com/ (accessed on 14 November 2019).
- Freedman, B.S.; Lam, A.Q.; Sundsbak, J.L.; Iatrino, R.; Su, X.; Koon, S.J.; Wu, M.; Daheron, L.; Harris, P.C.; Zhou, J.; et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J. Am. Soc. Nephrol. 2013, 24, 1571–1586. [Google Scholar] [CrossRef] [Green Version]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [Green Version]
- Cruz, N.M.; Song, X.; Czerniecki, S.M.; Gulieva, R.E.; Churchill, A.J.; Kim, Y.K.; Winston, K.; Tran, L.M.; Diaz, M.A.; Fu, H.; et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater. 2017, 16, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Ameku, T.; Taura, D.; Sone, M.; Numata, T.; Nakamura, M.; Shiota, F.; Toyoda, T.; Matsui, S.; Araoka, T.; Yasuno, T.; et al. Identification of MMP1 as a novel risk factor for intracranial aneurysms in ADPKD using iPSC models. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ashammakhi, N.; Wesseling-Perry, K.; Hasan, A.; Elkhammas, E.; Zhang, Y.S. Kidney-on-a-chip: Untapped opportunities. Kidney Int. 2018, 94, 1073–1086. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nichols, C.D. Human Disease Models in Drosophila melanogaster and the Role of the Fly in Therapeutic Drug Discovery. Pharm. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [Green Version]
- Strange, K. Drug Discovery in Fish, Flies, and Worms. ILAR J. 2016, 57, 133–143. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Directive 2010/63/EU Directive 2010/63/EU of the European Parliament and of the Council on the Protection of Animals Used for Scientific Purposes; European Commission: Brussels, Belgium, 2010. [Google Scholar]
- Schmitt, S.M.; Gull, M.; Brändli, A.W. Engineering Xenopus embryos for phenotypic drug discovery screening. Adv. Drug Deliv. Rev. 2014, 69–70, 225–246. [Google Scholar] [CrossRef]
- Barr, M.M.; Sternberg, P.W. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 1999, 401, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Barr, M.M.; DeModena, J.; Braun, D.; Nguyen, C.Q.; Hall, D.H.; Sternberg, P.W. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr. Biol. 2001, 11, 1341–1346. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.J.; Sharma, M. Polycystic Kidney Disease: Lessons Learned from Caenorhabditis elegans Mating Behavior. Curr. Biol. 2015, 25, R1168–R1170. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.U.; Zank, S.; Fabretti, F.; Benzing, T. Caenorhabditis elegans, a model organism for kidney research: From cilia to mechanosensation and longevity. Curr. Opin. Nephrol. Hypertens. 2011, 20, 400–408. [Google Scholar] [CrossRef]
- Wang, J.; Barr, M.M. Ciliary Extracellular Vesicles: Txt Msg Organelles. Cell. Mol. Neurobiol. 2016, 36, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Barr, M.M. Caenorhabditis elegans as a model to study renal development and disease: Sexy cilia. J. Am. Soc. Nephrol. 2005, 16, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Barr, M.M. ATP-2 Interacts with the PLAT Domain of LOV-1 and Is Involved in Caenorhabditis elegans Polycystin Signaling. Mol. Biol. Cell 2005, 16, 458–469. [Google Scholar] [CrossRef]
- Bae, Y.-K.; Lyman-Gingerich, J.; Barr, M.M.; Knobel, K.M. Identification of genes involved in the ciliary trafficking of C. elegans PKD-2. Dev. Dyn. 2008, 237, 2021–2029. [Google Scholar] [CrossRef] [Green Version]
- Knobel, K.M.; Peden, E.M.; Barr, M.M. Distinct protein domains regulate ciliary targeting and function of C. elegans PKD-2. Exp. Cell Res. 2008, 314, 825–833. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.-K.; Kim, E.; L’Hernault, S.W.; Barr, M.M. The CIL-1 PI 5-Phosphatase Localizes TRP Polycystins to Cilia and Activates Sperm in C. elegans. Curr. Biol. 2009, 19, 1599–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kaletsky, R.; Silva, M.; Williams, A.; Haas, L.; Androwski, R.; Landis, J.; Patrick, C.; Rashid, A.; Santiago-Martinez, D.; et al. Cell-specific transcriptional profiling of ciliated sensory neurons reveals regulators of behavior and extracellular vesicle biogenesis. Curr. Biol. 2015, 25, 3232–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peden, E.M.; Barr, M.M. The KLP-6 kinesin is required for male mating behaviors and polycystin localization in Caenorhabditis elegans. Curr. Biol. 2005, 15, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.M.; Portman, D.S. A latent capacity of the C. elegans polycystins to disrupt sensory transduction is repressed by the single-pass ciliary membrane protein CWP-5. Dis. Model. Mech. 2010, 3, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Barr, M.M.; Using, C. elegans as a model in PKD. In Polycystic Kidney Disease; Hu, J., Yu, Y., Eds.; CRC Press: New York, NY, USA, 2019; pp. 247–266. ISBN 9780429468834. [Google Scholar]
- Watnick, T.J.; Jin, Y.; Matunis, E.; Kernan, M.J.; Montell, C. A flagellar polycystin-2 homolog required for male fertility in Drosophila. Curr. Biol. 2003, 13, 2179–2184. [Google Scholar] [CrossRef]
- Köttgen, M.; Hofherr, A.; Li, W.; Chu, K.; Cook, S.; Montell, C.; Watnick, T. Drosophila Sperm Swim Backwards in the Female Reproductive Tract and Are Activated via TRPP2 Ion Channels. PLoS ONE 2011, 6, e20031. [Google Scholar] [CrossRef] [Green Version]
- Hofherr, A.; Wagner, C.J.; Watnick, T.; Köttgen, M. Targeted rescue of a polycystic kidney disease mutation by lysosomal inhibition. Kidney Int. 2016, 89, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Venglarik, C.J.; Gao, Z.; Lu, X. Evolutionary Conservation of Drosophilia Polycystin-2 as a Calcium-Activated Cation Channel. J. Am. Soc. Nephrol. 2004, 13, 2508–2516. [Google Scholar]
- Gamberi, C.; Hipfner, D.R.; Trudel, M.; Lubell, W.D. Bicaudal C mutation causes myc and TOR pathway up-regulation and polycystic kidney disease-like phenotypes in Drosophila. PLoS Genet. 2017, 13, e1006694. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Kurashige, M.; Liu, Y.; Terabayashi, T.; Ishimoto, Y.; Wang, T.; Choudhary, V.; Hobbs, R.; Liu, L.-K.; Lee, P.-H.; et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci. Rep. 2018, 8, 2743. [Google Scholar] [CrossRef]
- Vassilev, P.M.; Guo, L.; Chen, X.-Z.; Segal, Y.; Peng, J.-B.; Basora, N.; Babakhanlou, H.; Cruger, G.; Kanazirska, M.; Ye, C.; et al. Polycystin-2 Is a Novel Cation Channel Implicated in Defective Intracellular Ca2+ Homeostasis in Polycystic Kidney Disease. Biochem. Biophys. Res. Commun. 2001, 282, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Vandorpe, D.H.; Chernova, M.N.; Jiang, L.; Sellin, L.K.; Wilhelm, S.; Stuart-Tilley, A.K.; Walz, G.; Alper, S.L. The cytoplasmic C-terminal fragment of polycystin-1 regulates a Ca2+-permeable cation channel. J. Biol. Chem. 2001, 276, 4093–4101. [Google Scholar] [CrossRef] [Green Version]
- Chernova, M.N.; Vandorpe, D.H.; Clark, J.S.; Alper, S.L. Expression of the polycystin-1 C-terminal cytoplasmic tail increases Cl- channel activity inXenopus oocytes. Kidney Int. 2005, 68, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Streets, A.J.; Wessely, O.; Peters, D.J.M.; Ong, A.C.M. Hyperphosphorylation of polycystin-2 at a critical residue in disease reveals an essential role for polycystin-1-regulated dephosphorylation. Hum. Mol. Genet. 2013, 22, 1924–1939. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Streets, A.J.; Hounslow, A.M.; Tran, U.; Jean-Alphonse, F.; Needham, A.J.; Vilardaga, J.-P.; Wessely, O.; Williamson, M.P.; Ong, A.C.M. The Polycystin-1, Lipoxygenase, and α-Toxin Domain Regulates Polycystin-1 Trafficking. J. Am. Soc. Nephrol. 2016, 27, 1159–1173. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca2+ signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef]
- Zhang, B.; Tran, U.; Wessely, O. Polycystin 1 loss of function is directly linked to an imbalance in G-protein signaling in the kidney. Development 2018, 145, dev158931. [Google Scholar] [CrossRef] [Green Version]
- Tran, U.; Zakin, L.; Schweickert, A.; Agrawal, R.; Döger, R.; Blum, M.; De Robertis, E.M.; Wessely, O. The RNA-binding protein bicaudal C regulates polycystin 2 in the kidney by antagonizing miR-17 activity. Development 2010, 137, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Tran, U.; Pickney, L.M.; Ozpolat, B.D.; Wessely, O. Xenopus Bicaudal-C is required for the differentiation of the amphibian pronephros. Dev. Biol. 2007, 307, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Drummond, I.A. Kidney development and disease in the zebrafish. J. Am. Soc. Nephrol. 2005, 16, 299–304. [Google Scholar] [CrossRef]
- Gehring, T.V.; Luksys, G.; Sandi, C.; Vasilaki, E. Detailed classification of swimming paths in the Morris Water Maze: Multiple strategies within one trial. Sci. Rep. 2015, 5, 14562. [Google Scholar] [CrossRef]
- Kim, E.; Arnould, T.; Sellin, L.K.; Benzing, T.; Fan, M.J.; Grüning, W.; Sokol, S.Y.; Drummond, I.; Walz, G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem. 1999, 274, 4947–4953. [Google Scholar] [CrossRef] [Green Version]
- Low, S.H.; Vasanth, S.; Larson, C.H.; Mukherjee, S.; Sharma, N.; Kinter, M.T.; Kane, M.E.; Obara, T.; Weimbs, T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell 2006, 10, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Amsterdam, A.; Pazour, G.J.; Cole, D.G.; Miller, M.S.; Hopkins, N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development 2004, 127, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Bouvrette, D.J.; Sittaramane, V.; Heidel, J.R.; Chandrasekhar, A.; Bryda, E.C. Knockdown of bicaudal C in zebrafish (Danio rerio) causes cystic kidneys: A nonmammalian model of polycystic kidney disease. Comp. Med. 2010, 60, 96–106. [Google Scholar]
- Sullivan-Brown, J.; Schottenfeld, J.; Okabe, N.; Hostetter, C.L.; Serluca, F.C.; Thiberge, S.Y.; Burdine, R.D. Zebrafish mutations affecting cilia motility share similar cystic phenotypes and suggest a mechanism of cyst formation that differs from pkd2 morphants. Dev. Biol. 2008, 314, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Obara, T.; Mangos, S.; Liu, Y.; Zhao, J.; Wiessner, S.; Kramer-Zucker, A.G.; Olale, F.; Schier, A.F.; Drummond, I.A. Polycystin-2 Immunolocalization and Function in Zebrafish. J. Am. Soc. Nephrol. 2006, 17, 2706–2718. [Google Scholar] [CrossRef] [Green Version]
- Schottenfeld, J.; Sullivan-Brown, J.; Burdine, R.D. Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific-expression of southpaw. Development 2007, 127, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Pennekamp, P.; Karcher, C.; Fischer, A.; Schweickert, A.; Skryabin, B.; Horst, J.; Blum, M.; Dworniczak, B. The Ion Channel Polycystin-2 Is Required for Left-Right Axis Determination in Mice. Curr. Biol. 2002, 12, 938–943. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Wang, Y.; Schetle, N.; Gao, H.; Pütz, M.; Von Gersdorff, G.; Walz, G.; Kramer-Zucker, A.G. The subcellular localization of TRPP2 modulates its function. J. Am. Soc. Nephrol. 2008, 19, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Tietz Bogert, P.S.; Huang, B.Q.; Gradilone, S.A.; Masyuk, T.V.; Moulder, G.L.; Ekker, S.C.; Larusso, N.F. The zebrafish as a model to study polycystic liver disease. Zebrafish 2013, 10, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, C.R.; Ward, C.J.; Leightner, A.C.; Smith, J.L.; Agarwal, R.; Harris, P.C.; Torres, V.E. Phosphodiesterase 1A modulates cystogenesis in zebrafish. J. Am. Soc. Nephrol. 2014, 25, 2222–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, Y. Approaches to Studying Polycystic Kidney Disease in Zebrafish. In Polycystic Kidney Disease; Hu, J., Yu, Y., Eds.; CRC Press: New York, NY, USA, 2019; pp. 267–292. ISBN 9780429468834. [Google Scholar]
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–399. [Google Scholar] [CrossRef]
- Ganner, A.; Neumann-Haefelin, E. Genetic kidney diseases: Caenorhabditis elegans as model system. Cell Tissue Res. 2017, 369, 105–118. [Google Scholar] [CrossRef]
- Millet-Boureima, C.; Porras Marroquin, J.; Gamberi, C. Modeling Renal Disease “On the Fly”. Biomed. Res. Int. 2018, 2018, 5697436. [Google Scholar] [CrossRef] [Green Version]
- Millet-Boureima, C.; Chingle, R.; Lubell, W.D.; Gamberi, C. Cyst Reduction in a Polycystic Kidney Disease Drosophila Model Using Smac Mimics. Biomedicines 2019, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Boletta, A. Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics 2009, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Swanhart, L.M.; Cosentino, C.C.; Diep, C.Q.; Davidson, A.J.; de Caestecker, M.; Hukriede, N.A. Zebrafish kidney development: Basic science to translational research. Birth Defects Res. Part C Embryo Today Rev. 2011, 93, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Kok, F.O.; Shin, M.; Gupta, A.; Grosse, A.S.; van Impel, A.; Peterson, -M.; Kourkoulis, G.; Male, I.; DeSantis, D.F.; Tindell, S.S.; et al. Reverse genetic screening reveals poor correlation between Morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Metzner, A.; Griffiths, J.D.; Streets, A.J.; Markham, E.; Philippou, T.; Van Eeden, F.J.M.; Ong, A.C.M. A high throughput zebrafish chemical screen reveals ALK5 and non-canonical androgen signalling as modulators of the pkd2 −kd phenotype. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Blackburn, A.T.M.; Miller, R.K. Modeling congenital kidney diseases in Xenopus laevis. Dis. Model. Mech. 2019, 12, dmm038604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Semanchik, N.; Lee, S.H.; Somlo, S.; Barbano, P.E.; Coifman, R.; Sun, Z. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc. Natl. Acad. Sci. USA 2009, 106, 21819–21824. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Gu, J.; Mei, S.; Xu, D.; Jing, Y.; Yao, Q.; Chen, M.; Yang, M.; Chen, S.; Yang, B.; et al. Resveratrol delays polycystic kidney disease progression through attenuation of nuclear factor κB-induced inflammation. Nephrol. Dial. Transpl. 2016, 31, 1826–1834. [Google Scholar] [CrossRef]
- Chang, M.-Y.; Ma, T.-L.; Hung, C.-C.; Tian, Y.-C.; Chen, Y.-C.; Yang, C.-W.; Cheng, Y.-C. Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci. Rep. 2017, 7, 7161. [Google Scholar] [CrossRef]
- Westhoff, J.H.; Giselbrecht, S.; Schmidts, M.; Schindler, S.; Beales, P.L.; Tönshoff, B.; Liebel, U.; Gehrig, J. Development of an Automated Imaging Pipeline for the Analysis of the Zebrafish Larval Kidney. PLoS ONE 2013, 8, e82137. [Google Scholar] [CrossRef] [Green Version]
- Bryda, E.C. The Mighty Mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211. [Google Scholar]
- Motenko, H.; Neuhauser, S.B.; O’Keefe, M.; Richardson, J.E. MouseMine: A new data warehouse for MGI. Mammalian Genome 2015, 26, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, B.S.; Majumdar, S.S. An Efficient Method for Generation of Transgenic Rats Avoiding Embryo Manipulation. Mol. Ther. Nucl. Acids 2016, 5, e293. [Google Scholar] [CrossRef] [Green Version]
- Menezes, L.F.; Germino, G.G. Murine Models of Polycystic Kidney Disease. Drug Discov. Today Dis. Mech. 2013, 10, e153–e158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guay-Woodford, L.M. Murine models of polycystic kidney disease: Molecular and therapeutic insights. Am. J. Physiol. Ren. Physiol. 2003, 285, 1034–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhya, P. Models of polycystic kidney disease. Methods Mol. Med. 2003, 86, 13–28. [Google Scholar] [PubMed]
- Torres, V.E.; Harris, P.C. Polycystic kidney disease: Genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J. Intern. Med. 2007, 261, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D. Chapter 6 Mouse Models of Polycystic Kidney Disease. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2008; Volume 84, pp. 311–350. ISBN 9780123744548. [Google Scholar]
- Ko, J.Y.; Park, J.H. Mouse models of polycystic kidney disease induced by defects of ciliary proteins. BMB Rep. 2013, 46, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Cogswell, C.; Price, S.J.; Hou, X.; Guay-Woodford, L.M.; Flaherty, L.; Bryda, E.C. Positional cloning of jcpk/bpk locus of the mouse. Mammalian Genome 2003, 14, 242–249. [Google Scholar]
- Lehman, J.M.; Michaud, E.J.; Schoeb, T.R.; Aydin-Son, Y.; Miller, M.; Yoder, B.K. The Oak Ridge Polycystic Kidney mouse: Modeling ciliopathies of mice and men. Dev. Dyn. 2008, 237, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Hiesberger, T.; Cordes, K.; Sinclair, A.M.; Goldstein, L.S.B.; Somlo, S. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2003, 100, 5286–5291. [Google Scholar] [CrossRef] [Green Version]
- Trudel, M.; D’Agati, V.; Costantini, F. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int. 1991, 39, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Trudel, M.; Barisoni, L.; Lanoix, J.; D’Agati, V. Polycystic kidney disease in SBM transgenic mice: Role of c-myc in disease induction and progression. Am. J. Pathol. 1998, 152, 219–229. [Google Scholar]
- Flaherty, L.; Bryda, E.C.; Collins, D.; Rudofsky, U.; Montgomery, J.C. New mouse model for polycystic kidney disease with both recessive and dominant gene effects. Kidney Int. 1995, 47, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Atala, A.; Freeman, M.R.; Mandell, J.; Beier, D.R. Juvenile cystic kidneys (jck): A new mouse mutation which causes polycystic kidneys. Kidney Int. 1993, 43, 1081–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Lu, W.; Obara, T.; Kuida, S.; Lehoczky, J.; Dewar, K.; Drummond, I.A.; Beier, D.R. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002, 129, 5839–5846. [Google Scholar] [CrossRef] [Green Version]
- Janaswami, P.M.; Birkenmeier, E.H.; Cook, S.A.; Rowe, L.B.; Bronson, R.T.; Davisson, M.T. Identification and Genetic Mapping of a New Polycystic Kidney Disease on Mouse Chromosome 8. Genomics 1997, 40, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, P.; Birkenmeier, E.H.; Birkenmeier, C.S.; Barker, J.E. Mutations in a NIMA-related kinase gene, Nek1, cause pleiotropic effects including a progressive polycystic kidney disease in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Calvet, J.P.; Dittemore-Hoover, D.; Yoshida, K.; Grantham, J.J.; Gattone, V.H. A hereditary model of slowly progressive polycystic kidney disease in the mouse. J. Am. Soc. Nephrol. 1991, 1, 980–989. [Google Scholar]
- Olbrich, H.; Fliegauf, M.; Hoefele, J.; Kispert, A.; Otto, E.; Volz, A.; Wolf, M.T.; Sasmaz, G.; Trauer, U.; Reinhardt, R.; et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003, 34, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Kaspareit-Rittinghausen, J.; Wcislo, A. Animal model of human disease. Hereditary Polycystic Kidney Disease. Am. J. Pathol. 1991, 139, 693–696. [Google Scholar]
- Cowley, B.D.; Gudapaty, S.; Kraybill, A.L.; Barash, B.D.; Harding, M.A.; Calvet, J.P.; Gattone, V.H. Autosomal-dominant polycystic kidney disease in the rat. Kidney Int. 1993, 43, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Bihoreau, M.; Ceccherini, I.; Browne, J.; Kränzlin, B.; Romeo, G.; Lathrop, G.M.; James, M.R.; Gretz, N. Location of the first genetic locus, PKDr1, controlling autosomal dominant polycystic kidney disease in Han:SPRD cy/+ rat. Hum. Mol. Genet. 1997, 6, 609–613. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.H.; Bihoreau, M.-T.; Hoffmann, S.; Kränzlin, B.; Tychinskaya, I.; Obermüller, N.; Podlich, D.; Boehn, S.N.; Kaisaki, P.J.; Megel, N.; et al. Missense Mutation in Sterile α Motif of Novel Protein SamCystin is Associated with Polycystic Kidney Disease in ( cy/+) Rat. J. Am. Soc. Nephrol. 2005, 16, 3517–3526. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, P.; Somlo, S. Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2384–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; D’Agati, V.; Cai, Y.; Markowitz, G.; Park, J.H.; Reynolds, D.M.; Maeda, Y.; Le, T.C.; Hou, H.; Kucherlapati, R.; et al. Somatic Inactivation of Pkd2 Results in Polycystic Kidney Disease. Cell 1998, 93, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Markowitz, G.S.; Li, L.; D’Agati, V.D.; Factor, S.M.; Geng, L.; Tibara, S.; Tuchman, J.; Cai, Y.; Park, J.H.; et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat. Genet. 2000, 24, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Leeuwen, I.S.L.; Dauwerse, J.G.; Baelde, H.J.; Leonhard, W.N.; van de Wal, A.; Ward, C.J.; Verbeek, S.; DeRuiter, M.C.; Breuning, M.H.; de Heer, E.; et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 2004, 13, 3069–3077. [Google Scholar] [CrossRef] [Green Version]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [Green Version]
- Hopp, K.; Hommerding, C.J.; Wang, X.; Ye, H.; Harris, P.C.; Torres, V.E. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J. Am. Soc. Nephrol. 2015, 26, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Kurbegovic, A.; Côté, O.; Couillard, M.; Ward, C.J.; Harris, P.C.; Trudel, M. Pkd1 transgenic mice: Adult model of polycystic kidney disease with extrarenal and renal phenotypes. Hum. Mol. Genet. 2010, 19, 1174–1189. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.J.; Turley, H.; Ong, A.C.M.; Comley, M.; Biddolph, S.; Chetty, R.; Ratcliffe, P.J.; Gattner, K.; Harris, P.C. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc. Natl. Acad. Sci. USA 1996, 93, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhao, J.; Jiang, W.J.; Shan, X.W.; Yang, X.M.; Gao, J.G. Conditional gene manipulation: Cre-ating a new biological era. J. Zhejiang Univ. Sci. B 2012, 13, 511–524. [Google Scholar] [CrossRef]
- Shibazaki, S.; Yu, Z.; Nishio, S.; Tian, X.; Thomson, R.B.; Mitobe, M.; Louvi, A.; Velazquez, H.; Ishibe, S.; Cantley, L.G.; et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 2008, 17, 1505–1516. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Somlo, S.; Igarashi, P. Epithelial-Specific Cre/lox Recombination in the Developing Kidney and Genitourinary Tract. J. Am. Soc. Nephrol. 2002, 13, 1837–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piontek, K.; Menezes, L.F.; Garcia-Gonzalez, M.A.; Huso, D.L.; Germino, G.G. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat. Med. 2007, 13, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Holditch, S.J.; Nemenoff, R.A.; Hopp, K. Rodent Autosomal Dominant Polycystic Kidney Disease Models. In Polycystic Kidney Disease; Hu, J., Yu, Y., Eds.; CRC Press: New York, NY, USA, 2019; pp. 195–246. ISBN 9780429468834. [Google Scholar]
- Schieren, G.; Pey, R.; Bach, J.; Hafner, M.; Gretz, N. Murine models of polycystic kidney disease. Nephrol. Dial. Transpl. 1996, 11, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biller, D.S.; DiBartola, S.P.; Eaton, K.A.; Pflueger, S.; Wellman, M.L.; Radin, M.J. Inheritance of Polycystic Kidney Disease in Persian Cats. J. Hered. 1996, 87, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Eaton, K.A.; Biller, D.S.; DiBartola, S.P.; Radin, M.J.; Wellman, M.L. Autosomal Dominant Polycystic Kidney Disease in Persian and Persian-cross Cats. Vet. Pathol. 1997, 34, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.M.; Pedersen, H.D.; Haggstrom, J.; Koch, J.; Ersbøll, A.K. Increased Mean Arterial Pressure and Aldosterone-to-Renin Ratio in Persian Cats with Polycystic Kidney Disease. J. Vet. Intern. Med. 2003, 17, 21–27. [Google Scholar] [CrossRef]
- Lyons, L.A.; Biller, D.S.; Erdman, C.A.; Lipinski, M.J.; Young, A.E.; Roe, B.A.; Qin, B.; Grahn, R.A. Feline polycystic kidney disease mutation identified in PKD1. J. Am. Soc. Nephrol. 2004, 15, 2548–2555. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Mrug, M.; Lyons, L.A.; Weimbs, T.; Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; et al. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023. [Google Scholar] [CrossRef]
- Gutierrez, K.; Dicks, N.; Glanzner, W.G.; Agellon, L.B.; Bordignon, V. Efficacy of the porcine species in biomedical research. Front. Genet. 2015, 6, 293. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, Z.; Busch, J.; Awwad, M.; Wagner, R.; Wells, K.; Cooper, D.K.C. Selected physiologic compatibilities and incompatibilities between human and porcine organ systems. Xenotransplantation 2006, 13, 488–499. [Google Scholar] [CrossRef]
- He, J.; Wang, Q.; Ye, J.; Hu, X.; Li, N. Identification of porcine polycystic kidney disease 1 (PKD1) gene: Molecular cloning, expression profile, and implication in disease model. Gene 2011, 490, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yin, H.; He, J.; Ye, J.; Ding, F.; Wang, S.; Hu, X.; Meng, Q.; Li, N. cDNA cloning of porcine PKD2 gene and RNA interference in LLC–PK1 cells. Gene 2011, 476, 38–45. [Google Scholar] [CrossRef]
- He, J.; Li, Q.; Fang, S.; Guo, Y.; Liu, T.; Ye, J.; Yu, Z.; Zhang, R.; Zhao, Y.; Hu, X.; et al. Pkd1 mono-allelic knockout is sufficient to trigger renal cystogenesis in a mini-pig model. Int. J. Biol. Sci. 2015, 11, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, X.; Wu, X.; Li, Z.; Zhang, Y.; Song, K.; Cai, G.; Li, Q.; Lin, S.; Chen, X.; Bai, X.Y. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br. J. Pharm. 2019, 176, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; He, J.; Li, Q.; Feng, Y.; Bai, X.; Chen, X.; Zhao, Y.; Hu, X.; Yu, Z.; Li, N. Generation of c-Myc transgenic pigs for autosomal dominant polycystic kidney disease. Transgenic Res. 2013, 22, 1231–1239. [Google Scholar] [CrossRef]
- He, J.; Ye, J.; Li, Q.; Feng, Y.; Bai, X.; Chen, X.; Wu, C.; Yu, Z.; Zhao, Y.; Hu, X.; et al. Construction of a transgenic pig model overexpressing polycystic kidney disease 2 (PKD2) gene. Transgenic Res. 2013, 22, 861–867. [Google Scholar] [CrossRef]
- Zhang, L.; Vijg, J. Somatic Mutagenesis in Mammals and Its Implications for Human Disease and Aging. Annu. Rev. Genet. 2018, 52, 397–419. [Google Scholar] [CrossRef]
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin Markedly Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wüthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han: SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transpl. 2006, 21, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Wahl, P.R.; Le Hir, M.; Wäckerle-Men, Y.; Wüthrich, R.P.; Serra, A.L. Everolimus Retards Cyst Growth and Preserves Kidney Function in a Rodent Model for Polycystic Kidney Disease. Kidney Blood Press. Res. 2007, 30, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and Kidney Growth in Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Walz, G.; Budde, K.; Mannaa, M.; Nürnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Gattone, V.H.; Wang, X.; Harris, P.C.; Torres, V.E. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003, 9, 1323–1326. [Google Scholar] [CrossRef]
- Torres, V.E.; Wang, X.; Qian, Q.; Somlo, S.; Harris, P.C.; Gattone, V.H. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004, 10, 363–364. [Google Scholar] [CrossRef]
- Wang, X.; Gattone, V.; Harris, P.C.; Torres, V.E. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J. Am. Soc. Nephrol. 2005, 16, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Meijer, E.; Gansevoort, R.T.; De Jong, P.E.; Van Der Wal, A.M.; Leonhard, W.N.; De Krey, S.R.; Van Den Born, J.; Mulder, G.M.; Van Goor, H.; Struck, J.; et al. Therapeutic potential of vasopressin V2 receptor antagonist in a mouse model for autosomal dominant polycystic kidney disease: Optimal timing and dosing of the drug. Nephrol. Dial. Transpl. 2011, 26, 2445–2453. [Google Scholar] [CrossRef] [Green Version]
- Gattone, V.H.; Maser, R.L.; Tian, C.; Rosenberg, J.M.; Branden, M.G. Developmental expression of urine concentration-associated genes and their altered expression in murine infantile-type polycystic kidney disease. Dev. Genet. 1999, 24, 309–318. [Google Scholar] [CrossRef]
- Sullivan, J.; Karra, K.; Moxon, S.A.T.; Vallejos, A.; Motenko, H.; Wong, J.D.; Aleksic, J.; Balakrishnan, R.; Binkley, G.; Harris, T.; et al. InterMOD: Integrated data and tools for the unification of model organism research. Sci. Rep. 2013, 3, 1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]




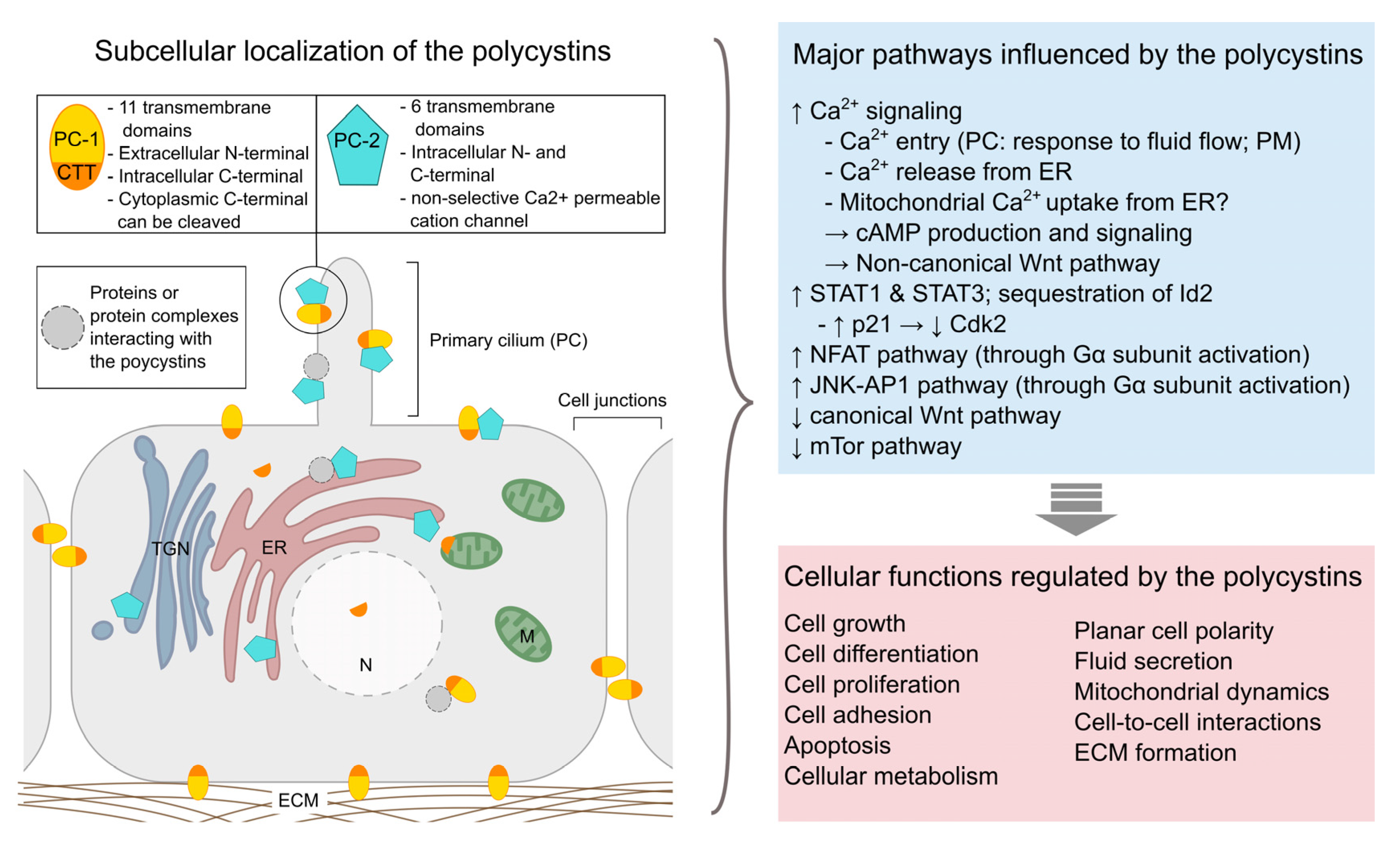{kind=link}
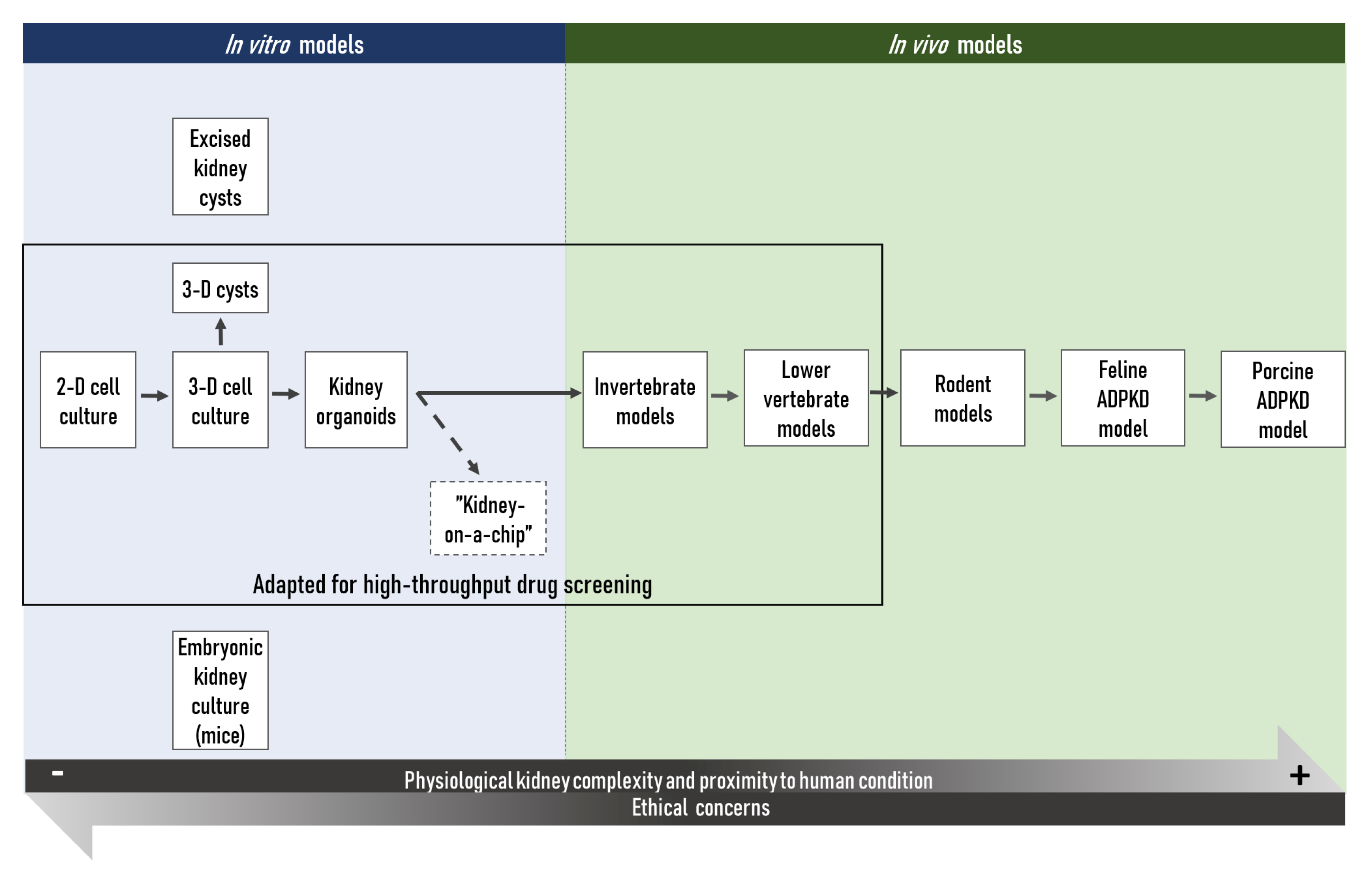{kind=link}
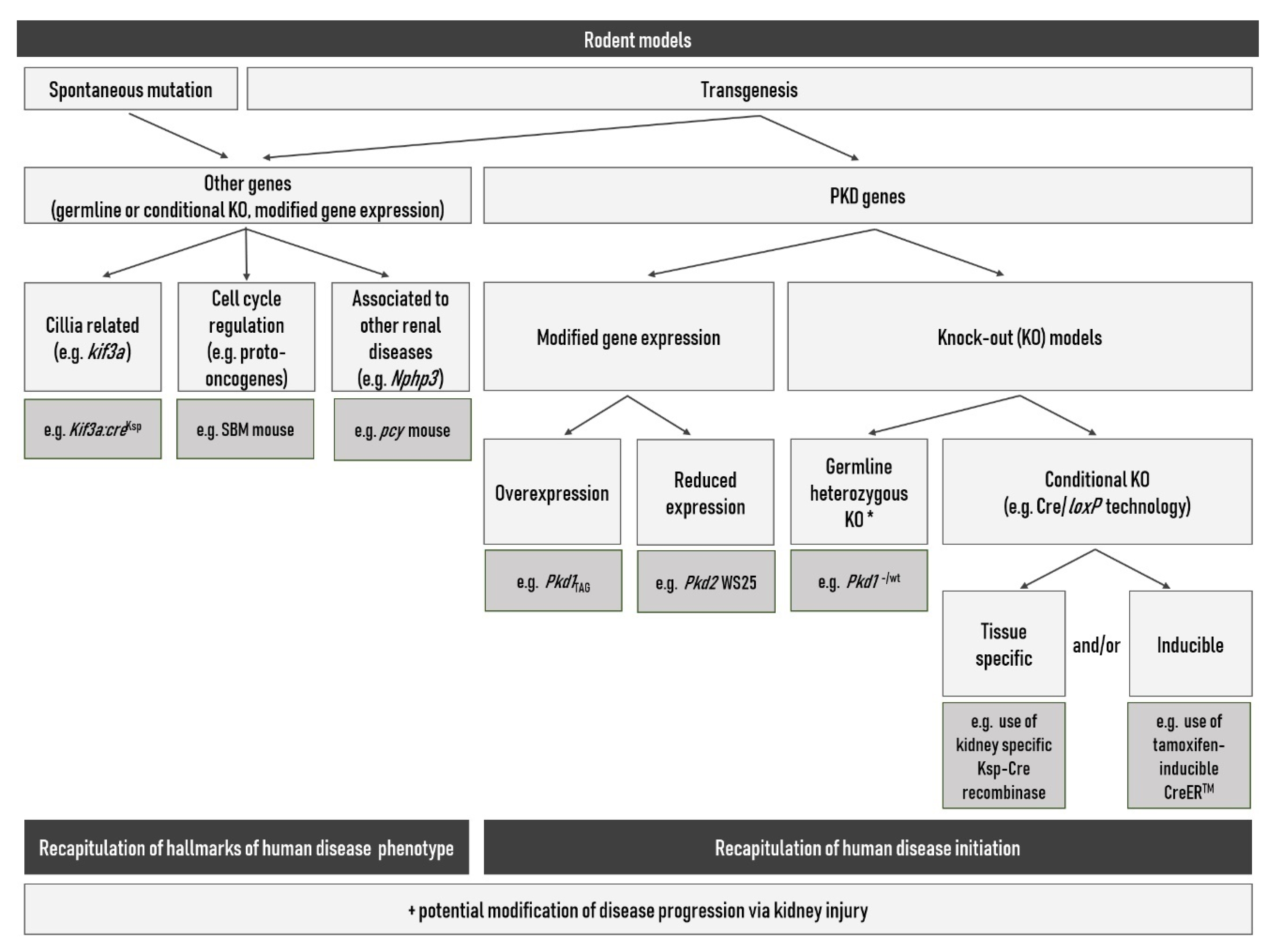{kind=link}
| Advantages | Limitations | Ref. | |
|---|---|---|---|
| Cell Type | |||
| Primary cells |
|
| [33,34] |
| Immortalized cell lines |
|
| [33,34,35] |
| Pluripotent stem cells (PSC) |
|
| [32,33,34,36,37] |
| Culture System | |||
| 2D cell culture |
|
| [34,38] |
| Spheroid culture (3D cysts) |
|
| [38,39] |
| Embryonic kidney culture (rodents) |
|
| [40,41] |
| Kidney organoids |
|
| [32,33,36,37] |
| Origin of Cells | |||
| Human |
|
| [33,34,39] |
| Animal |
|
| [34,39] |
| Species | Excretory System | Advantages | Limitations | Applications (ADPKD) | Ref. |
|---|---|---|---|---|---|
| Invertebrates | |||||
| C. elegans | Rudimentary kidney-like organ consisting of a single cell |
|
|
| [85,92,101,131,132] |
| D. melanogaster | Aglomerular, Malpighian tubules (analog to renal tubules) and nephrocytes (analog to podocytes) |
|
|
| [84,85,133,134,135] |
| Vertebrates | |||||
| D. rerio | Pronephros with two nephrons (embryos), mesonephros (adults) |
|
|
| [22,121,123,128,130,136,137,138] |
| Xenopus (X. laevisand X. tropicalis) | Pronephros with two nephrons (embryos), mesonephros (adults) |
|
|
| [87,139] |
| Example | Gene | Construct/Mutation | Human Orthologue | Protein | Phenotype | Ref. | |
|---|---|---|---|---|---|---|---|
| Induced mutation (cilia associated gene) | Kif3a:creKsp | kif3a | Kidney specific inactivation | KIF3A | KIF3A (subunit of kinesin-II) | Renal parenchyma replaced with cysts by postnatal day 35 | [155] |
| Induced mutation (proto-onco gene) | SBM mouse | c-myc | c-myc overexpression via the introduction of a transgene (c-myc coding region under the control of the SV40 enhancer and β-globin promoter, different lines with variable copy number of the transgene) | MIC | Myc proto-ongogene protein | Progressive polycystic kidney disease with atypical plasmacytic infiltrates, anemia, premature death due to renal failure at 2 weeks to 4 months of age | [156,157] |
| Spontaneous mutation | jcpk mouse | Bicc1 | Single base-pair change (AG→AA) in the splice acceptor site of exon 3 causing a frameshift resulting in a premature stop codon | BICC1 | Bicc1 | Progressive polycystic kidney disease, hepatic and pancreatic dilated ducts, gall bladder enlargement, premature death between P7-P10 | [153,158] |
| jck mouse | Nek8 | Nucleotide substitution (G→T) resulting in an amino acid change (V→G) | NEK8 | Nek8 (NIMA (never in mitosis-A) related kinase) | Slowly progressive polycystic kidney disease, decreased fertility in males from 15 weeks of age, premature death at 20 to 25 weeks of age | [159,160] | |
| kat and kat2J mouse | Nek1 | kat: internal deletion kat2J: single base-pair insertion causing a frame shift resulting in a premature stop codon | NEK1 | Nek1 (NIMA (never in mitosis-A) related kinase) | Late onset, slow-progressing polycystic kidney disease, facial dysmorphism, dwarfing, male sterility, anemia, and cystic choroid plexus, premature death either before weaning or at 1 year of age (faster disease progression in kat2J) | [161,162] | |
| pcy mouse | Nphp3 | Amino acid substitution (I→S) | NPHP3 (associated with nephronoph-thisis) | Nephrocystin-3 | Slow progressive polycystic kidney disease with cerebral aneurysms, premature death and chronic inflammatory infiltrates in advanced stages, with a mean age at death of 6.5 months (females) and 8.2 months (males) | [163,164] | |
| Han:SPRD cy/+ rat (Cy-rat) | Anks6 (Pkdr1) | Missense point mutation resulting in an amino acid substitution (R→W) | ANKS6 | Ankyrin repeat and SAM domain-containing protein 6 | Slowly progressing renal enlargement by cyst formation, phenotype more severe in males than in females, premature death (males from 6 months of age, later for females) | [165,166,167,168] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koslowski, S.; Latapy, C.; Auvray, P.; Blondel, M.; Meijer, L. An Overview of In Vivo and In Vitro Models for Autosomal Dominant Polycystic Kidney Disease: A Journey from 3D-Cysts to Mini-Pigs. Int. J. Mol. Sci. 2020, 21, 4537. https://doi.org/10.3390/ijms21124537
Koslowski S, Latapy C, Auvray P, Blondel M, Meijer L. An Overview of In Vivo and In Vitro Models for Autosomal Dominant Polycystic Kidney Disease: A Journey from 3D-Cysts to Mini-Pigs. International Journal of Molecular Sciences. 2020; 21(12):4537. https://doi.org/10.3390/ijms21124537
Chicago/Turabian StyleKoslowski, Svenja, Camille Latapy, Pierrïck Auvray, Marc Blondel, and Laurent Meijer. 2020. "An Overview of In Vivo and In Vitro Models for Autosomal Dominant Polycystic Kidney Disease: A Journey from 3D-Cysts to Mini-Pigs" International Journal of Molecular Sciences 21, no. 12: 4537. https://doi.org/10.3390/ijms21124537
APA StyleKoslowski, S., Latapy, C., Auvray, P., Blondel, M., & Meijer, L. (2020). An Overview of In Vivo and In Vitro Models for Autosomal Dominant Polycystic Kidney Disease: A Journey from 3D-Cysts to Mini-Pigs. International Journal of Molecular Sciences, 21(12), 4537. https://doi.org/10.3390/ijms21124537